
Polyoxazolines with a Vicinally Double-Bioactivated Terminus for Biomacromolecular Affinity Assessment

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
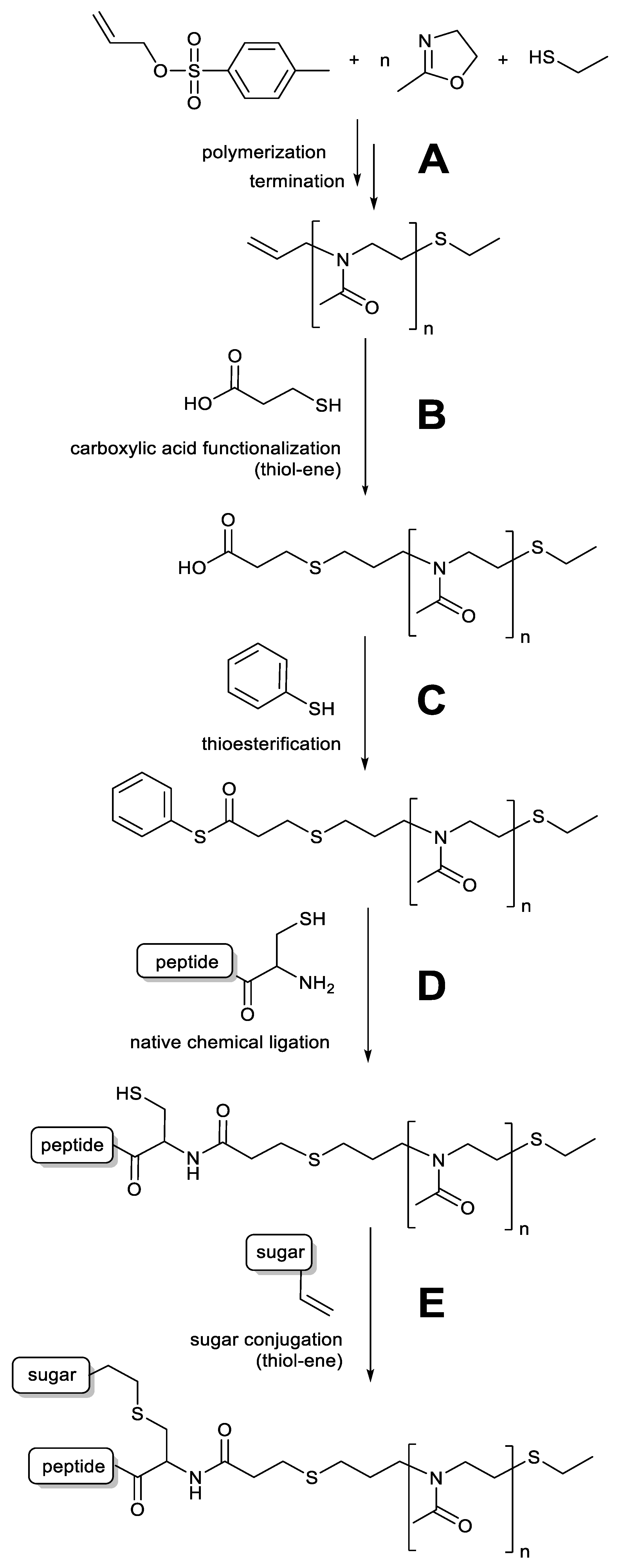
2.3. Synthesis
3. Results
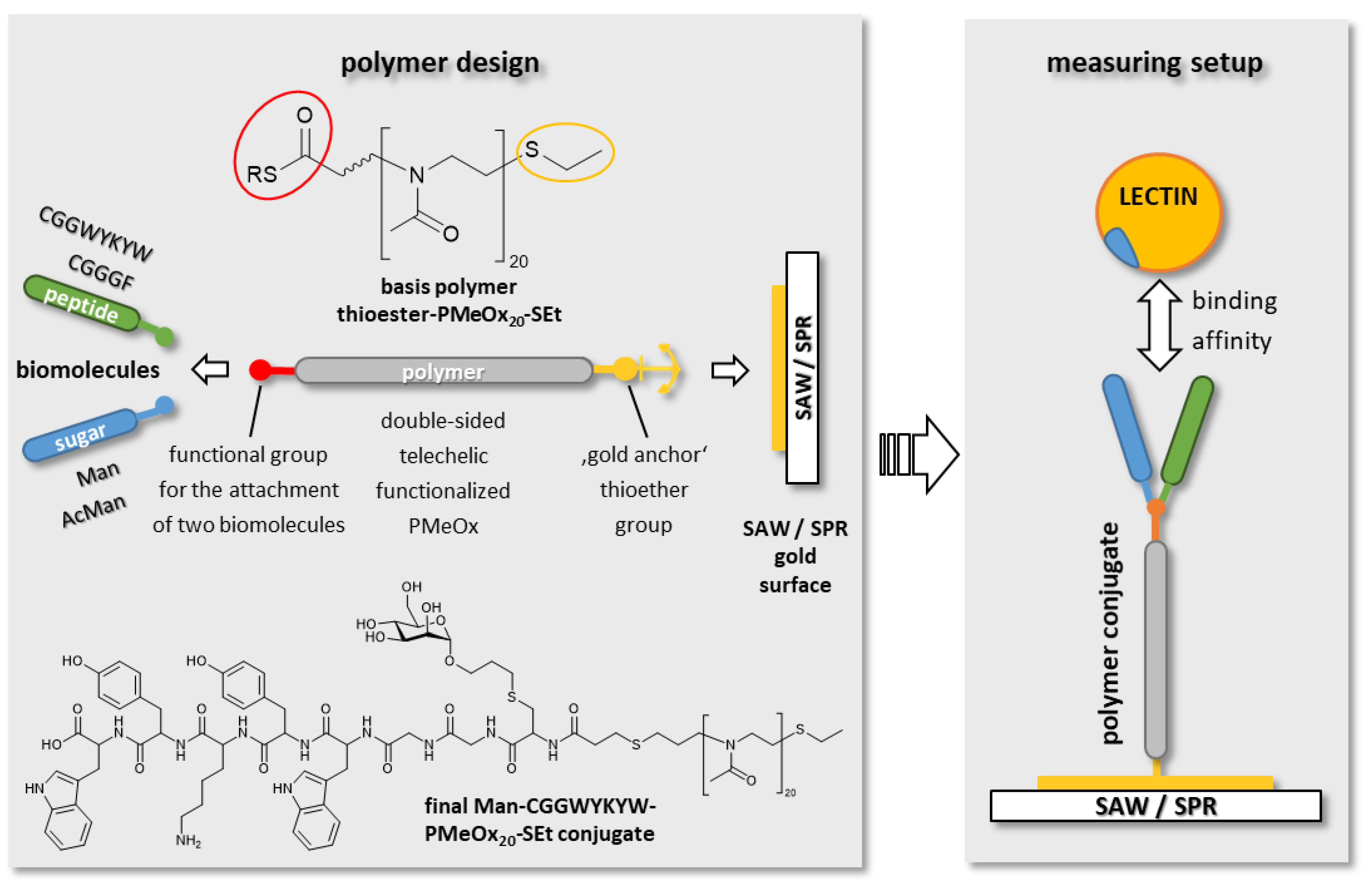
3.1. Overall Aim and Strategy
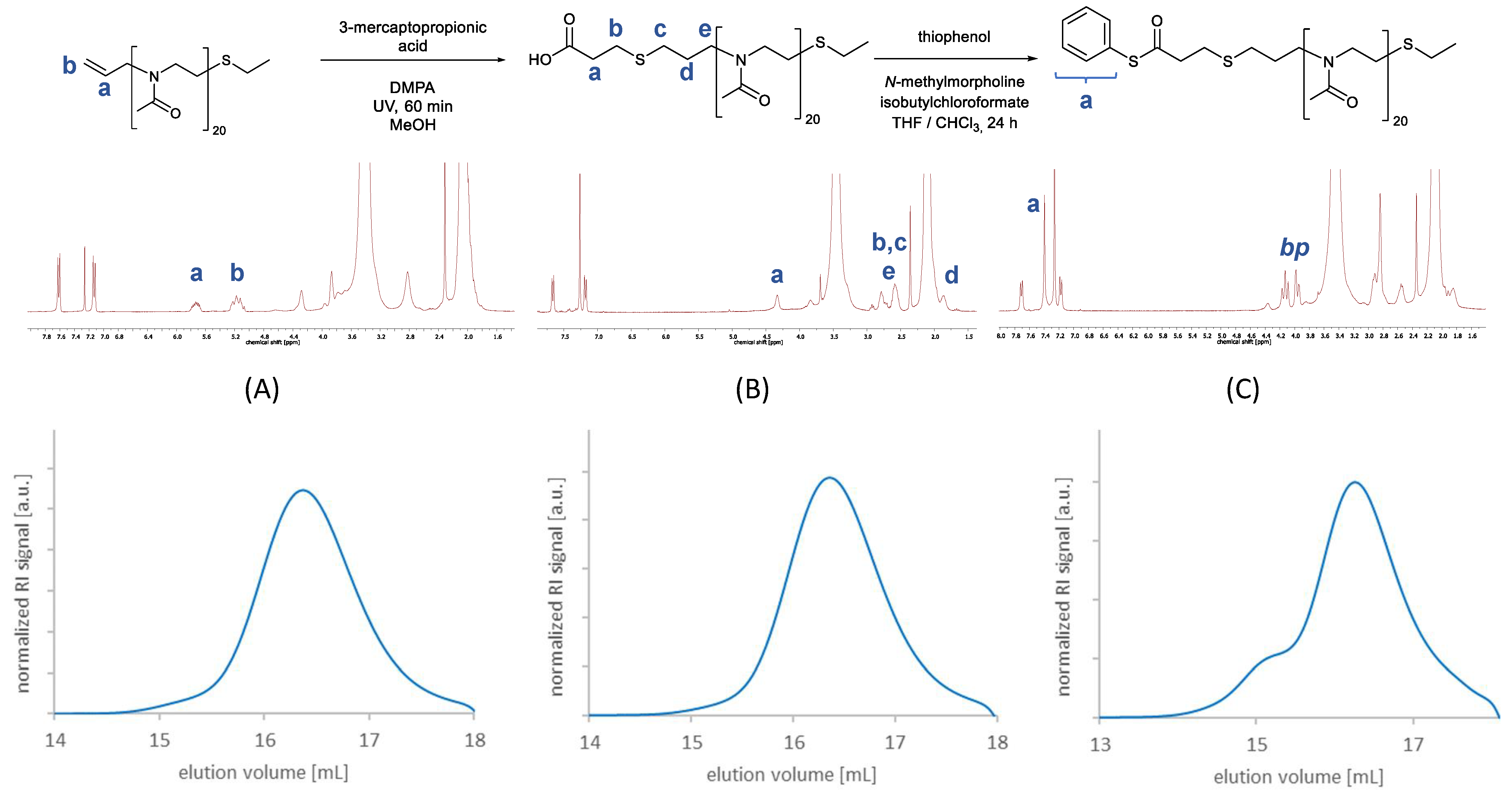
3.1.1. Synthesis of Functionalized Polyoxazolines
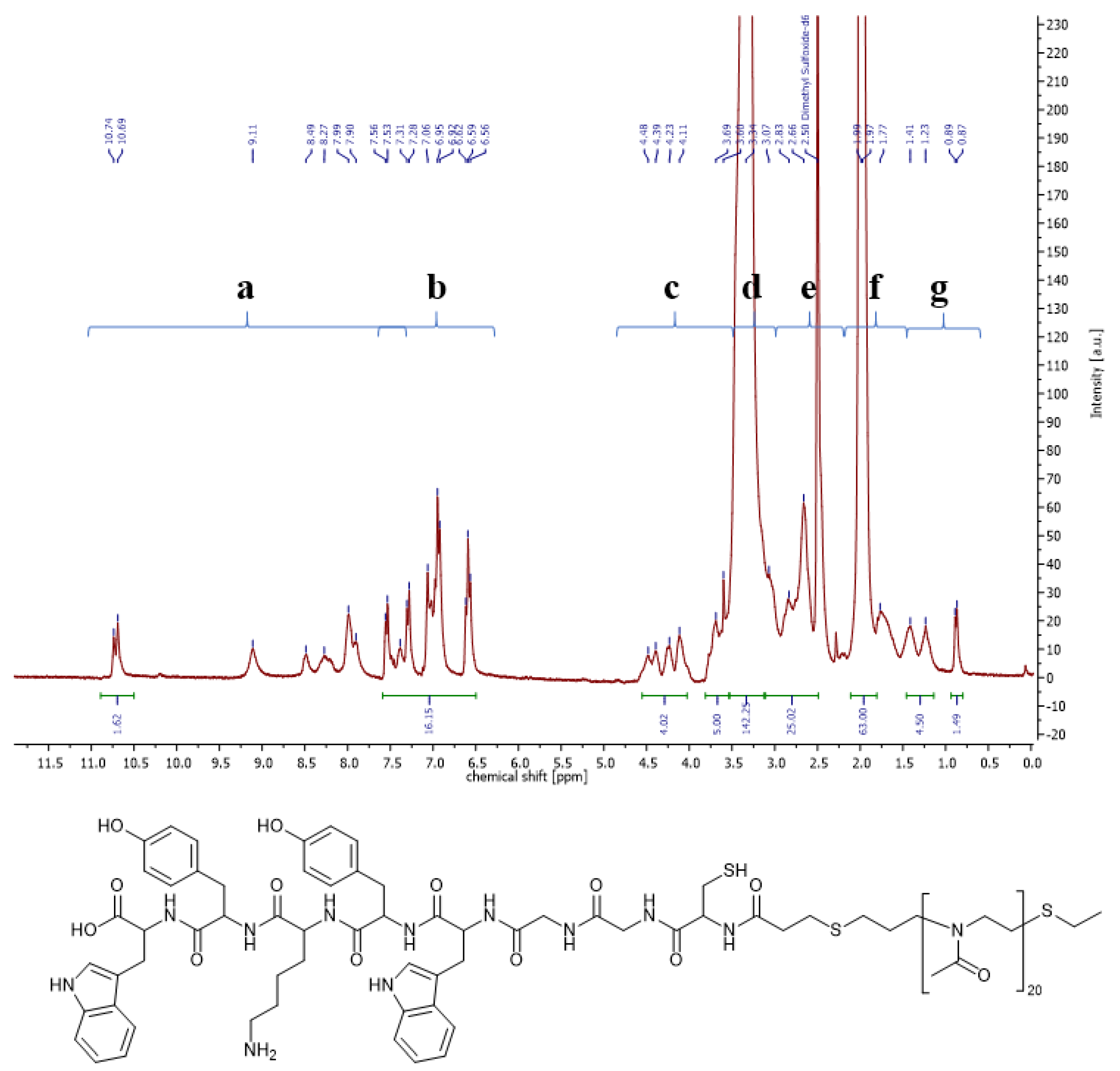
3.1.2. Synthesis of Two-Part Polymer Peptide Conjugates via NCL
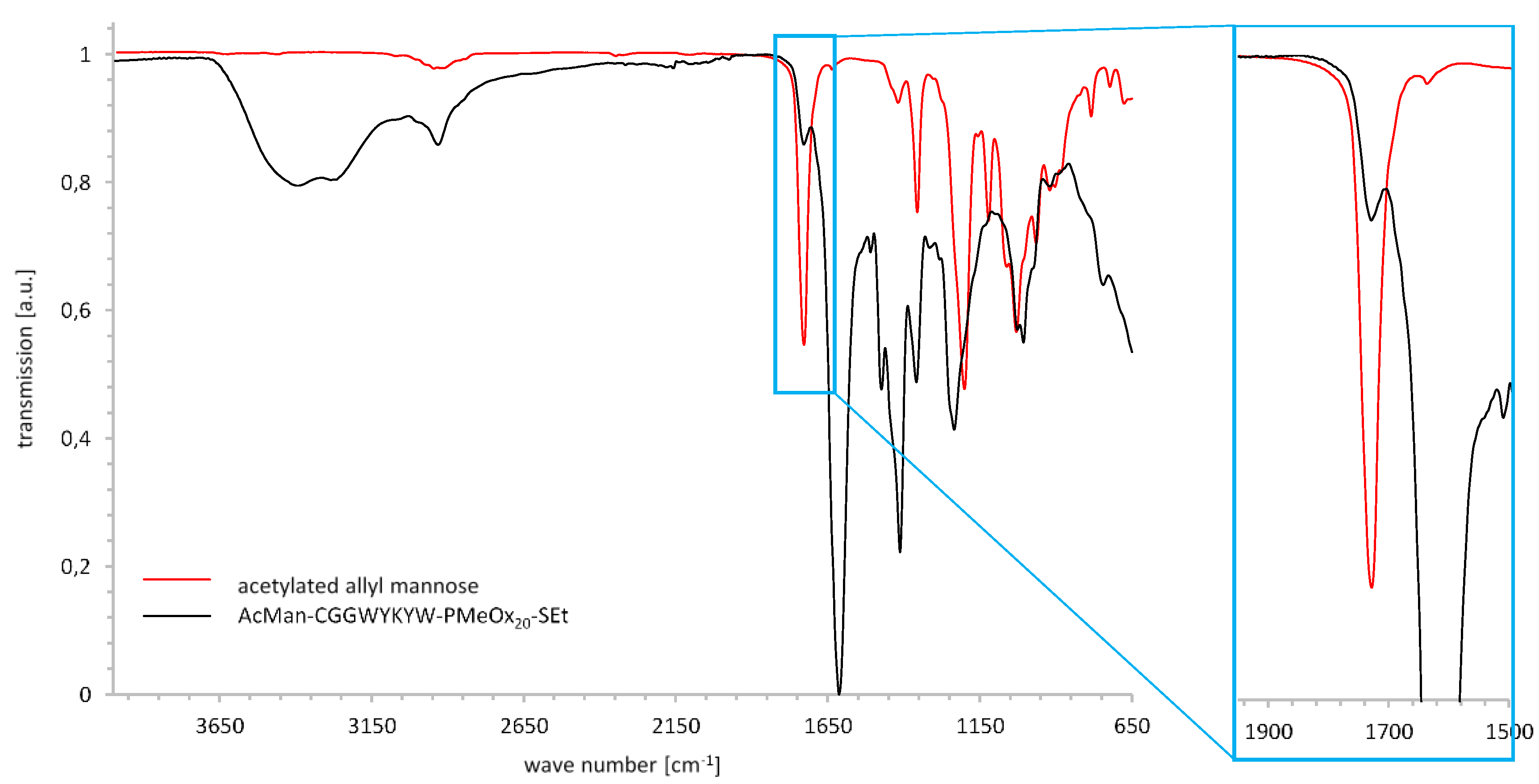
3.1.3. Synthesis of Three-Part Polymer Sugar Conjugates via Thiol-Ene Reaction
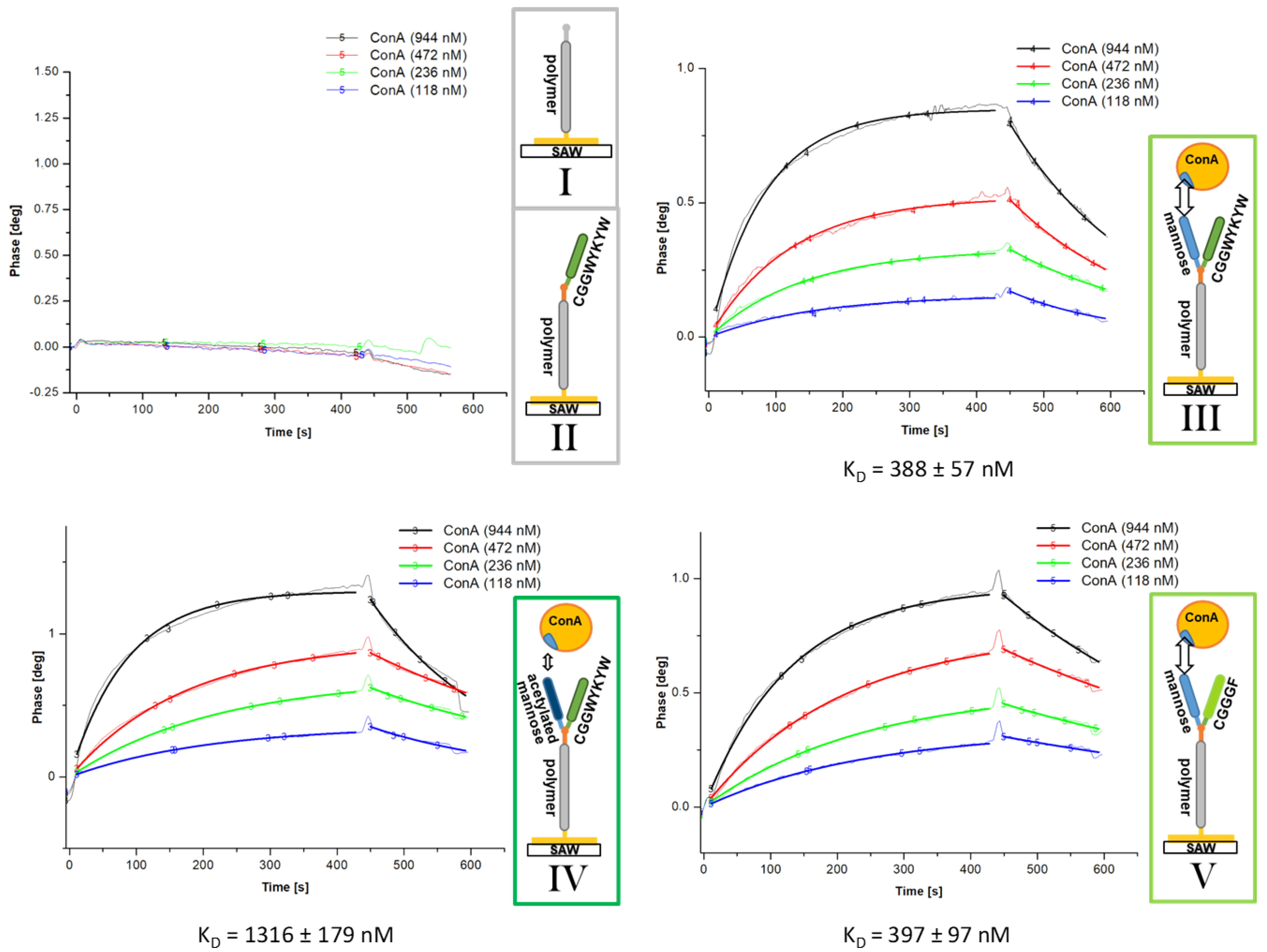
3.2. Biomacromolecular Recognition Experiments
SAW Affinity Studies
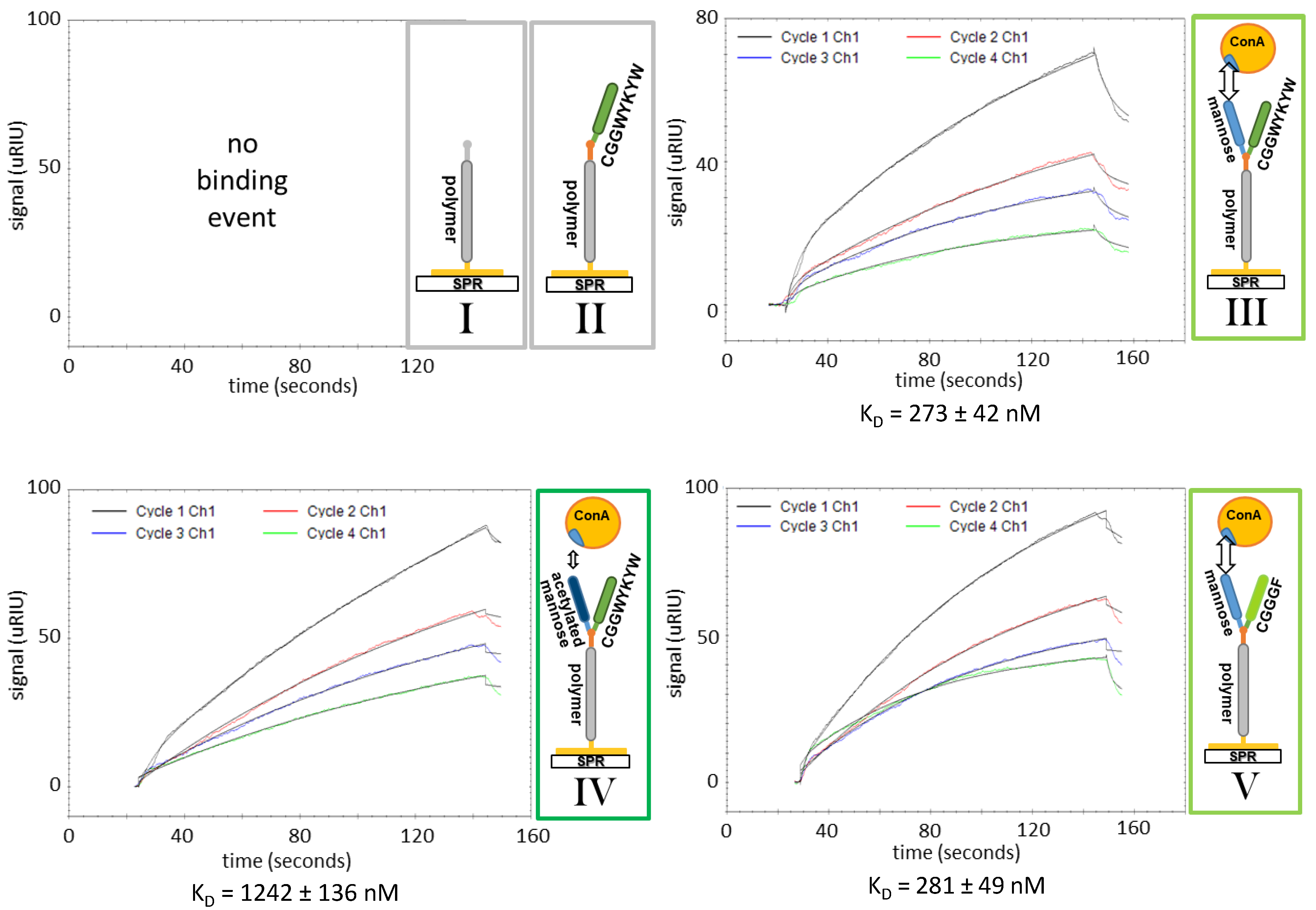
3.3. SPR Affinity Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water networks contribute to enthalpy/entropy compensation in protein–ligand binding. J. Am. Chem. Soc. 2013, 135, 15579–15584. [Google Scholar] [CrossRef] [Green Version]
- Simmel, F. Cellular and biomolecular recognition. synthetic and non-biological molecules. Angew. Chem. Int. Ed. 2010, 49, 1721. [Google Scholar] [CrossRef]
- Sharon, N.; Lis, H. Lectins as cell recognition molecules. Science 1989, 246, 227. [Google Scholar] [CrossRef]
- Whitesides, G.; Mammen, M.; Choi, S. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar]
- Muller, C.; Despras, G.; Lindhorst, T.K. Organizing multivalency in carbohydrate recognition. Chem. Soc. Rev. 2016, 45, 3275–3302. [Google Scholar] [CrossRef] [Green Version]
- Claes, D.; Memmel, E.; Holzapfel, M.; Seibel, J.; Maison, W. High-affinity carbohydrate binding by trimeric benzoboroxoles measured on carbohydrate arrays. ChemBioChem 2014, 15, 2450–2457. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, R.T.; Varki, A. Neoglycoconjugates: Preparation and applications. Trends Biochem. Sci. 1995, 20, 214. [Google Scholar]
- So, L.L.; Goldstein, I. Protein-carbohydrate interaction IV. application of the quantitative precipitin method to polysaccharide-concanavalin a interaction. J. Biol. Chem. 1967, 242, 1617–1622. [Google Scholar] [CrossRef]
- Vasu, K.S.; Naresh, K.; Bagul, R.S.; Jayaraman, N.; Sood, A.K. Detection of sugar-lectin interactions by multivalent dendritic sugar functionalized single-walled carbon nanotubes. Appl. Phys. Lett. 2012, 101, 053701. [Google Scholar] [CrossRef] [Green Version]
- Dam, T.K.; Roy, R.; Das, S.K.; Oscarson, S.; Brewer, C.F. Binding of multivalent carbohydrates to concanavalin a anddioclea grandiflora lectin THERMODYNAMIC ANALYSIS OF THE “MULTIVALENCY EFFECT”. J. Biol. Chem. 2000, 275, 14223–14230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.A.; Thomas, W.D.; Kiessling, L.L.; Corn, R.M. Surface plasmon resonance imaging studies of protein-carbohydrate interactions. J. Am. Chem. Soc. 2003, 125, 6140–6148. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, N.M.; Sampath, S.; Jayaraman, N. Synthesis and Langmuir studies of bivalent and monovalent α-D-mannopyranosides with Lectin Con A. Langmuir 2005, 21, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Ebara, Y.; Okahata, Y. A kinetic study of concanavalin A binding to glycolipid monolayers by using a quartz-crystal microbalance. J. Am. Chem. Soc. 1994, 116, 11209–11212. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, S.; Tang, Y.; Yu, L.; Hou, K.-Y.; Cheng, J.-P.; Zeng, X.; Wang, P.G. Carbohydrate− protein interactions by “clicked” carbohydrate self-assembled monolayers. Anal. Chem. 2006, 78, 2001–2008. [Google Scholar] [CrossRef]
- Laurent, N.; Haddoub, R.; Voglmeir, J.; Flitsch, S.L. MALDI-ToF MS analysis of glycosyltransferase activities on gold surface arrays. Methods Mol Biol. 2012, 808, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Yonzon, C.R.; Jeoung, E.; Zou, S.; Schatz, G.C.; Mrksich, M.; Van Duyne, R.P. A Comparative analysis of localized and propagating surface plasmon resonance sensors: The binding of concanavalin a to a monosaccharide functionalized self-assembled monolayer. J. Am. Chem. Soc. 2004, 126, 12669–12676. [Google Scholar] [CrossRef]
- Dechtrirat, D.; Gajovic-Eichelmann, N.; Bier, F.F.; Scheller, F.W. Hybrid material for protein sensing based on electrosynthesized mip on a mannose terminated self-assembled monolayer. Adv. Funct. Mater. 2014, 24, 2233–2239. [Google Scholar] [CrossRef]
- Abbina, S.; Parambath, A. 14-PEGylation and its alternatives: A summary. In Engineering of Biomaterials for Drug Delivery Systems; Parambath, A., Ed.; Woodhead Publishing: Cambridge, UK, 2018; pp. 363–376. [Google Scholar]
- Siegers, C.; Biesalski, M.; Haag, R. Self-assembled monolayers of dendritic polyglycerol derivatives on gold that resist the adsorption of proteins. Chem. Eur. J. 2004, 10, 2831–2838. [Google Scholar] [CrossRef]
- Barz, M.; Luxenhofer, R.; Zentel, R.; Vicent, M.J. Overcoming the PEG-addiction: Well-defined alternatives to PEG, from structure-property relationships to better defined therapeutics. Polym. Chem. 2011, 2, 1900–1918. [Google Scholar] [CrossRef]
- Grube, M.; Leiske, M.N.; Schubert, U.S.; Nischang, I. POx as an Alternative to PEG? a hydrodynamic and light scattering study. Macromolecules 2018, 51, 1905–1916. [Google Scholar] [CrossRef]
- van Hest, J.; Sumerlin, B.; Anseth, K.S.; Shoichet, M.; Hammes-Schiffer, S. Editorial for virtual issue on polymer bioconjugates in biology and medicine. Chem. Rev. 2017, 117, 900. [Google Scholar] [CrossRef] [Green Version]
- Reguera, L.; Méndez, Y.; Humpierre, A.R.; Valdés, O.; Rivera, D.G. Multicomponent reactions in ligation and bioconjugation chemistry. Acc. Chem. Res. 2018, 51, 1475–1486. [Google Scholar] [CrossRef] [PubMed]
- de Nooy, A.E.; Masci, G.; Crescenzi, V. Versatile synthesis of polysaccharide hydrogels using the Passerini and Ugi multicomponent condensations. Macromolecules 1999, 32, 1318–1320. [Google Scholar] [CrossRef]
- Ziegler, T.; Gerling, S.; Lang, M. Preparation of bioconjugates through an Ugi reaction. Angew. Chem. Int. Ed. 2000, 39, 2109–2112. [Google Scholar] [CrossRef]
- Méndez, Y.; Chang, J.; Humpierre, A.R.; Zanuy, A.; Garrido, R.; Vasco, A.V.; Pedroso, J.; Santana, D.; Rodríguez, L.M.; García-Rivera, D. Multicomponent polysaccharide–protein bioconjugation in the development of antibacterial glycoconjugate vaccine candidates. Chem. Sci. 2018, 9, 2581–2588. [Google Scholar] [CrossRef] [Green Version]
- Kreye, O.; Tóth, T.; Meier, M.A.R. Introducing multicomponent reactions to polymer science: Passerini reactions of renewable monomers. J. Am. Chem. Soc. 2011, 133, 1790–1792. [Google Scholar] [CrossRef]
- Driessen, F.; Martens, S.; De Meyer, B.; Du Prez, F.; Espeel, P. Double modification of polymer end groups through thiolactone chemistry. Macromol. Rapid Commun. 2016, 37, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Feineis, S.; Lutz, J.; Heffele, L.; Endl, E.; Albrecht, K.; Groll, J. Thioether–polyglycidol as multivalent and multifunctional coating system for gold nanoparticles. Adv. Mater. 2018, 30, 1704972. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Lee, T.Y.; Roper, T. Thiol–enes: Chemistry of the past with promise for the future. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5301–5338. [Google Scholar] [CrossRef]
- Kade, M.J.; Burke, D.J.; Hawker, C.J. The power of thiol-ene chemistry. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 743–750. [Google Scholar] [CrossRef]
- Chujo, Y.; Ihara, E.; lhara, H.; Saegusa, T. Synthesis of polysiloxane-polyoxazoline graft copolymer by hydrosilylation reaction. Polym. Bull. 1988, 19, 435–440. [Google Scholar] [CrossRef]
- Asano, K.; Matsubara, S. amphiphilic organocatalyst for schotten-baumann-type tosylation of alcohols under organic solvent free condition. Org. Lett. 2009, 11, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- MacDowell, D.W.; Purpura, J.M. Cope rearrangements in the thiophene series. J. Org. Chem. 1986, 51, 183–188. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Paulus, R.M.; Fijten, M.W.; Schubert, U.S. Concentration effects in the cationic ring-opening polymerization of 2-ethyl-2-oxazoline in N, N-dimethylacetamide. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1487–1497. [Google Scholar] [CrossRef]
- Litt, M.; Levy, A.; Herz, J. Polymerization of Cyclic Imino Ethers. X. Kinetics, Chain Transfer, and Repolymerization. J. Macromol. Sci. Part A Chem. 1975, 9, 703–727. [Google Scholar] [CrossRef]
- Monnery, B.D.; Jerca, V.V.; Sedlacek, O.; Verbraeken, B.; Cavill, R.; Hoogenboom, R. Defined high molar mass poly(2-Oxazoline)s. Angew. Chem. Int. Ed. 2018, 57, 15400–15404. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Monnery, B. Method for the Preparation of Uniform, High Molar Mass Cyclic Imino Ether Polymers. International Application No. PCT/EP2015/065829, 21 January 2016. [Google Scholar]
- Schmitz, M.; Kuhlmann, M.; Reimann, O.; Hackenberger, C.P.R.; Groll, J. Side-chain cysteine-functionalized poly(2-oxazoline)s for multiple peptide conjugation by native chemical ligation. Biomacromolecules 2015, 16, 1088–1094. [Google Scholar] [CrossRef]
- Markey, L.; Giordani, S.; Scanlan, E.M. Native chemical ligation, thiol–ene click: A methodology for the synthesis of functionalized peptides. J. Org. Chem. 2013, 78, 4270–4277. [Google Scholar] [CrossRef]
- Seidel, A. (Ed.) Characterization and Analysis of Polymers; Wiley-Interscience: Hoboken, NJ, USA, 2008. [Google Scholar]
- Line, B.R.; Mitra, A.; Nan, A.; Ghandehari, H. Targeting tumor angiogenesis: Comparison of peptide and polymer-peptide conjugates. J. Nucl. Med. 2005, 46, 1552–1560. [Google Scholar]
- Ting, S.S.; Chen, G.; Stenzel, M.H. Synthesis of glycopolymers and their multivalent recognitions with lectins. Polym. Chem. 2010, 1, 1392–1412. [Google Scholar] [CrossRef]
- Ambrosi, M.; Cameron, N.R.; Davis, B.G. Lectins: Tools for the molecular understanding of the glycocode. Org. Biomol. Chem. 2005, 3, 1593–1608. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAW KD [nM] | SPR KD [nM] | |||||||
|---|---|---|---|---|---|---|---|---|
| Lane 1 | Lane 2 | Lane 3 | Lane 1 | Lane 2 | Lane 3 | Lane 4 | ||
| allyl-PMeOx20-SEt (I) | no aff. | no aff. | ||||||
| CGGWYKYW-PMeOx20-SEt (II) | no aff. | no aff. | ||||||
| Man-CGGWYKYW-PMeOx20-SEt (III) | 351 ± 77 | 250 ± 34 | ||||||
| (1) | 257 ± 60 | 388 ± 92 | 383 ± 46 | 214 ± 32 | 229 ± 38 | 273 ± 42 | 285 ± 46 | |
| (2) | N/A | 329 ± 42 | 430 ± 34 | - | ||||
| (3) | 278 ± 44 | 432 ± 71 | 467 ± 62 | - | ||||
| (4) | 303 ± 75 | 388 ± 57 | 215 ± 25 | - | ||||
| AcMan-CGGWYKYW-PMeOx20-SEt (IV) | 1325 ± 97 | 1138 ± 193 | ||||||
| 1316 ± 179 | 1426 ± 293 | 1232 ± 266 | 894 ± 117 | 1081 ± 83 | 1242 ± 136 | 1336 ± 126 | ||
| Man-CGGGF-PMeOx20-SEt (V) | 332 ± 63 | 255 ± 40 | ||||||
| 272 ± 21 | 328 ± 55 | 397 ± 97 | 234 ± 41 | 204 ± 22 | 281 ± 49 | 293 ± 53 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinzner, F.; Keller, T.; Mut, J.; Bechold, J.; Seibel, J.; Groll, J. Polyoxazolines with a Vicinally Double-Bioactivated Terminus for Biomacromolecular Affinity Assessment. Sensors 2021, 21, 3153. https://doi.org/10.3390/s21093153
Pinzner F, Keller T, Mut J, Bechold J, Seibel J, Groll J. Polyoxazolines with a Vicinally Double-Bioactivated Terminus for Biomacromolecular Affinity Assessment. Sensors. 2021; 21(9):3153. https://doi.org/10.3390/s21093153
Chicago/Turabian StylePinzner, Florian, Thorsten Keller, Jürgen Mut, Julian Bechold, Jürgen Seibel, and Jürgen Groll. 2021. "Polyoxazolines with a Vicinally Double-Bioactivated Terminus for Biomacromolecular Affinity Assessment" Sensors 21, no. 9: 3153. https://doi.org/10.3390/s21093153