A Camera Sensors-Based System to Study Drug Effects on In Vitro Motility: The Case of PC-3 Prostate Cancer Cells

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
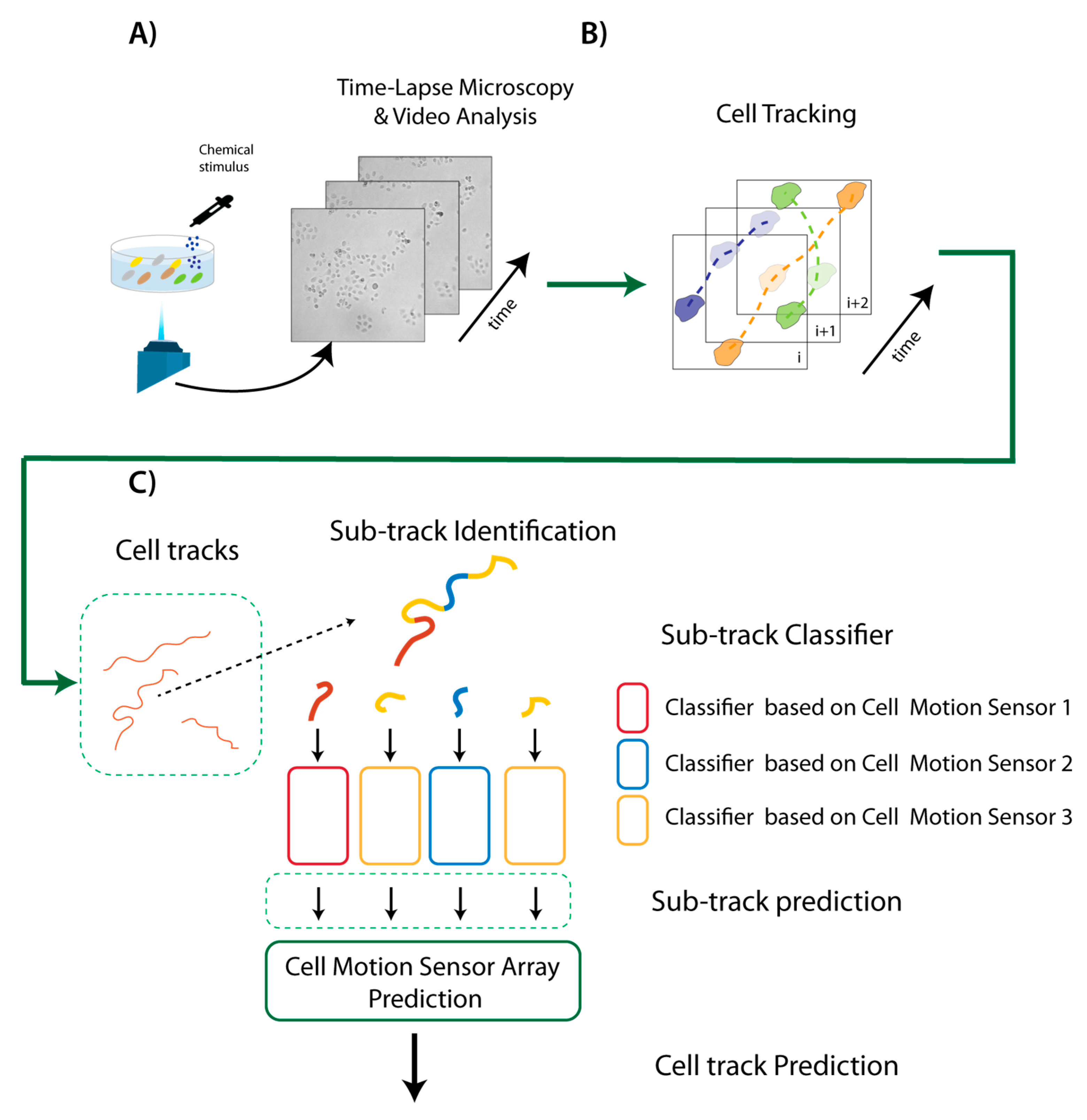
2.1. A Sketch of the Method
2.2. Cell Culture
2.3. Experimental Set-Up
2.4. Video Analysis
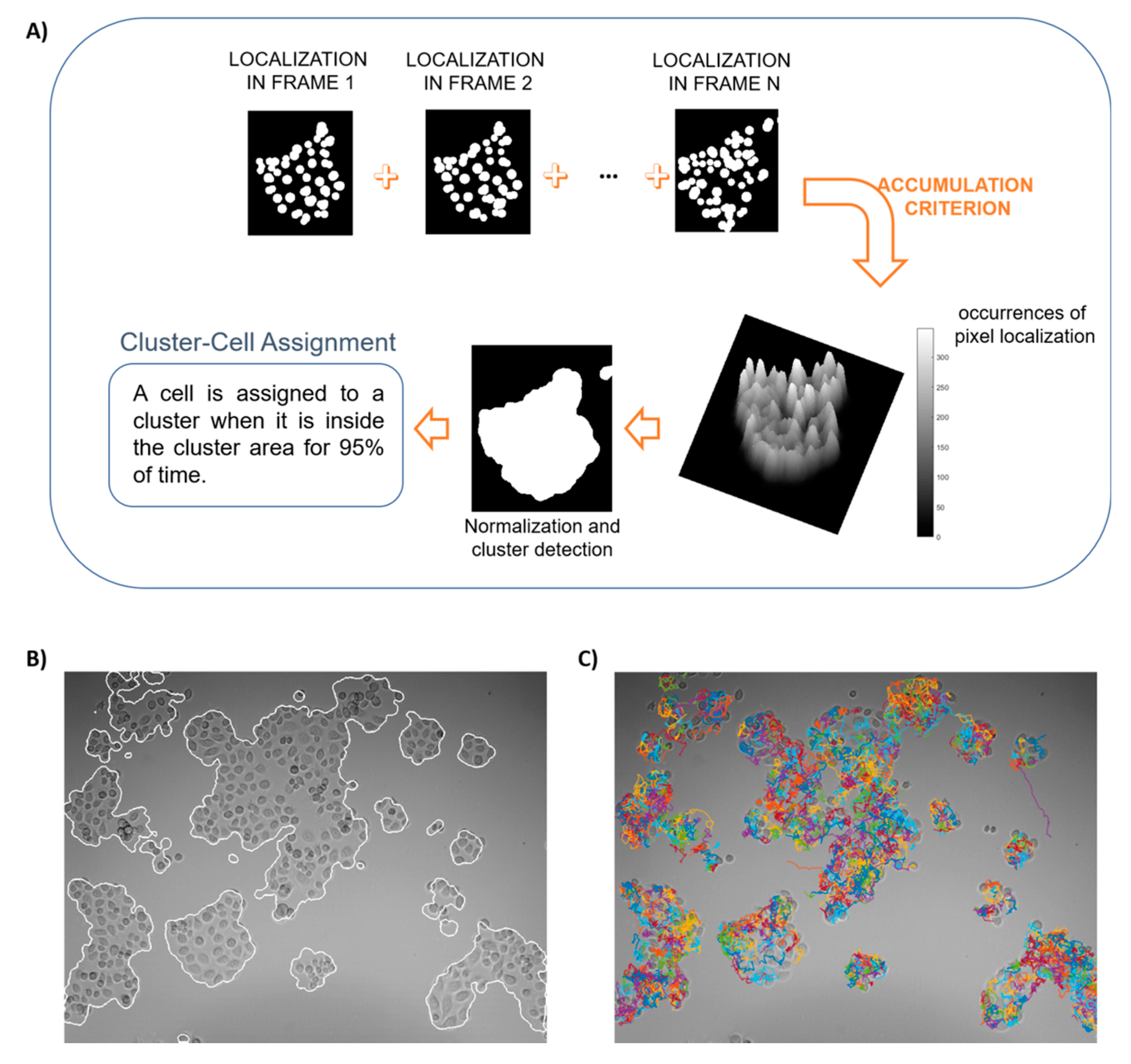
2.4.1. Automatic Clustering and Tracking
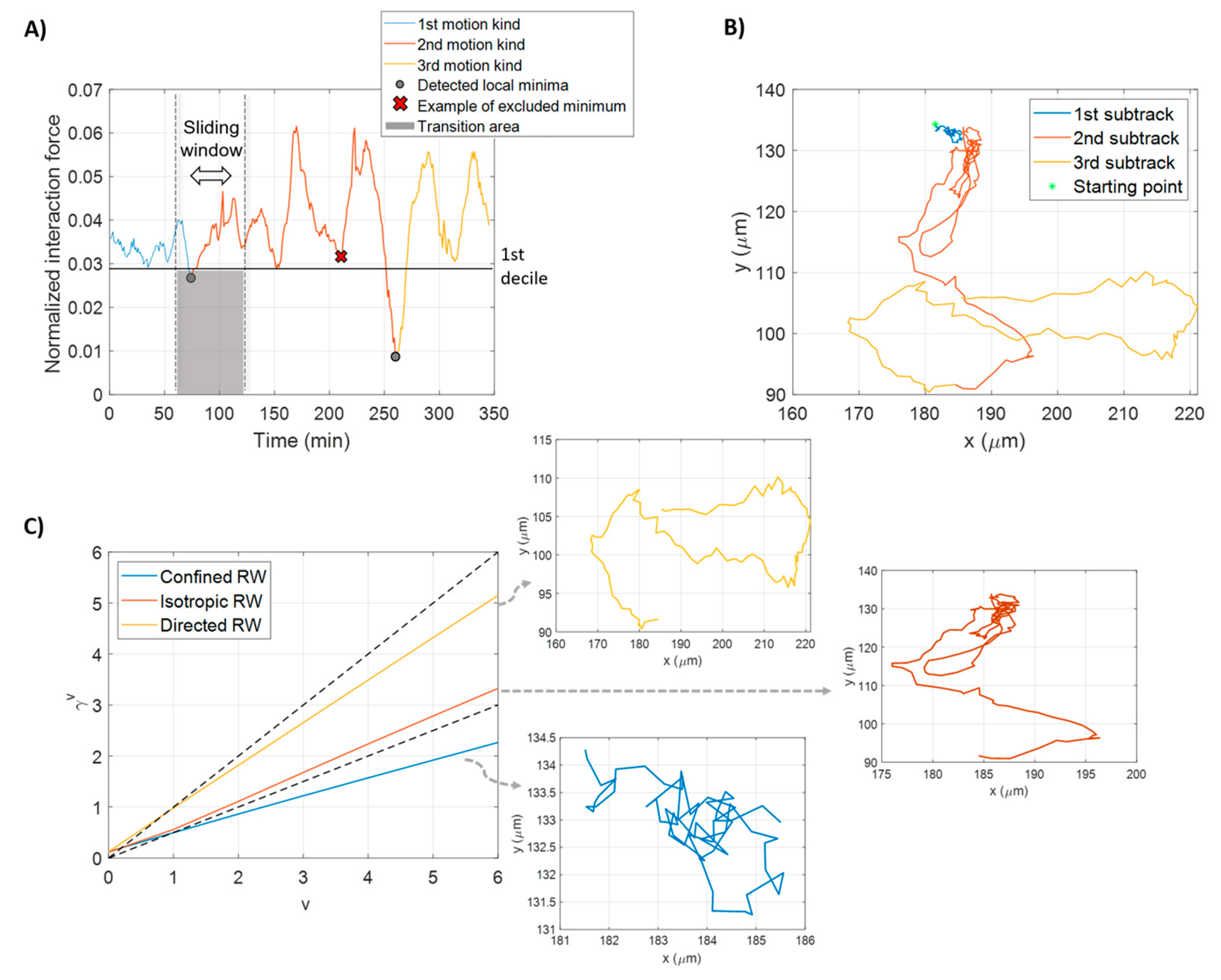
2.4.2. Sub-Track Identification within a Trajectory
2.4.3. Feature Extraction
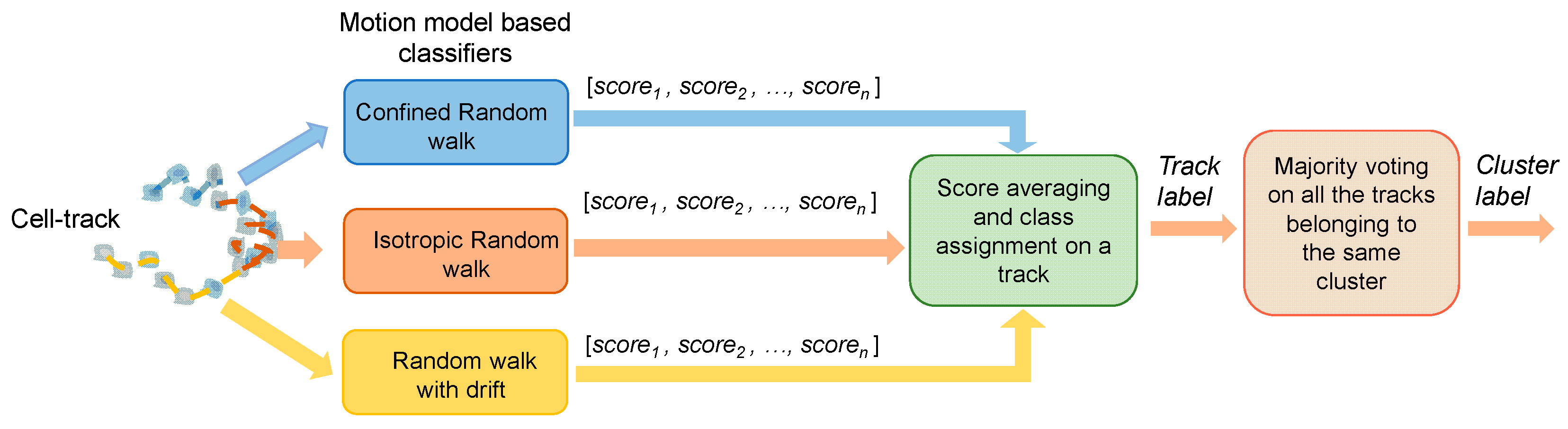
2.5. Machine Learning Architecture
2.5.1. Data Labeling
2.5.2. Good Teacher Sample Selection
2.5.3. Cooperative Strategies
3. Results and Discussion
3.1. Processed Data
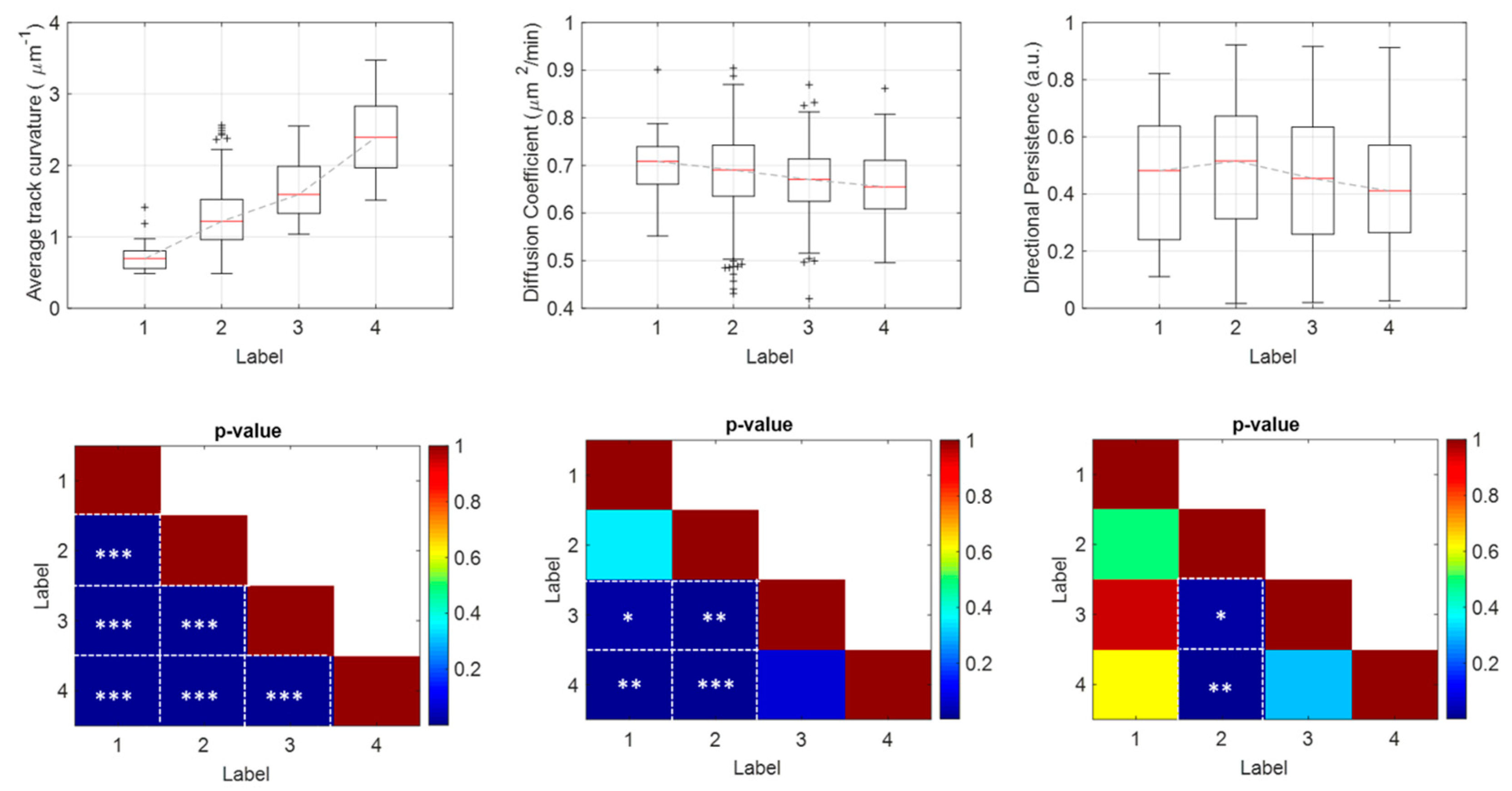
3.2. Beyond Univariate Data Analysis
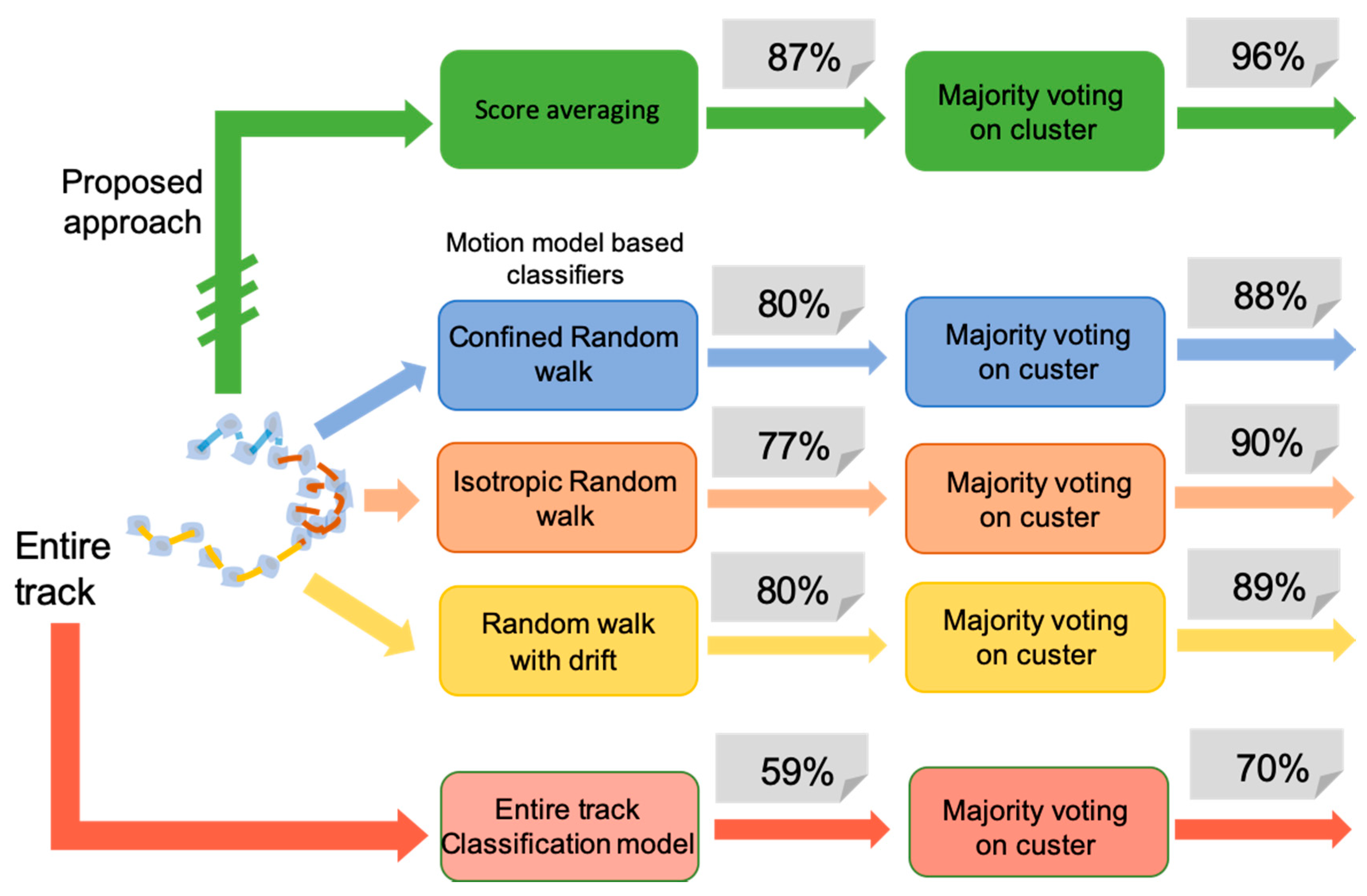
3.3. Comparative Approaches
3.4. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Collins, J.L.; Van Knippenberg, B.; Ding, K.; Kofman, A.V. Time-Lapse Microscopy. Cell Culture 2018, 3, 45–73. [Google Scholar]
- Ye, Y.; Liu, K.; Geng, S.; Ji, J.; Sun, J.; Zhang, Y.; Pi, F.; Sun, X. A novel fluorescent recombinant cell-based biosensor for screening NLRP3 inflammasome inhibitors. Sens. Actuators B Chem. 2019, 301. [Google Scholar] [CrossRef]
- Wei, X.; Gu, C.; Li, H.; Pan, Y.; Zhang, B.; Wang, P.; Wan, H.; Hu, N.; Wang, P. Efficacy and cardiotoxicity integrated assessment of anticancer drugs by a dual functional cell-based biosensor. Sens. Actuators B Chem. 2019, 283, 881–889. [Google Scholar] [CrossRef]
- Lee, S.H.; Jun, S.B.; Ko, H.J.; Kim, S.J.; Park, T.H. Cell-based olfactory biosensor using microfabricated planar electrode. Biosens. Bioelectron. 2009, 24, 2659–2664. [Google Scholar] [CrossRef]
- Oh, E.H.; Lee, S.H.; Lee, S.H.; Ko, H.J.; Park, T.H. Cell-based high-throughput odorant screening system through visualization on a microwell array. Biosens. Bioelectron. 2014, 53, 18–25. [Google Scholar] [CrossRef]
- Lee, S.H.; Oh, E.H.; Park, T.H. Cell-based microfluidic platform for mimicking human olfactory system. Biosens. Bioelectron. 2015, 74, 554–561. [Google Scholar] [CrossRef]
- Keefer, E.; Gramowski, A.; Stenger, D.; Pancrazio, J.; Gross, G. Characterization of acute neurotoxic effects of trimethylolpropane phosphate via neuronal network biosensors. Biosens. Bioelectron. 2001, 16, 513–525. [Google Scholar] [CrossRef]
- Ecken, H.; Ingebrandt, S.; Krause, M.; Richter, D.; Hara, M.; Offenhäusser, A. 64-Channel extended gate electrode arrays for extracellular signal recording. Electrochim. Acta 2003, 48, 3355–3362. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, C.; Cai, H.; Hu, N.; Zhou, J.; Wang, P. Cell-Based Biosensors and Their Application in Biomedicine. Chem. Rev. 2014, 114, 6423–6461. [Google Scholar] [CrossRef]
- Comes, M.C.; Casti, P.; Mencattini, A.; Di Giuseppe, D.; Mermet-Meillon, F.; De Ninno, A.; Parrini, M.C.; Businaro, L.; Di Natale, C.; Martinelli, E. The influence of spatial and temporal resolutions on the analysis of cell-cell interaction: A systematic study for time-lapse microscopy applications. Sci. Rep. 2019, 9, 6789. [Google Scholar] [CrossRef]
- Camley, B.A.; Zimmermann, J.; Levine, H.; Rappel, W.-J. Emergent collective chemotaxis without single-cell gradient sensing. Phys. Rev. Lett. 2016, 116. [Google Scholar] [CrossRef] [PubMed]
- Kabla, A. Collective cell migration: Leadership, invasion and segregation. J. R. Soc. Interface 2012, 9, 3268–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Boil. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Shaw, T.; Martin, P. Wound repair at a glance. J. Cell Sci. 2009, 122, 3209–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condeelis, J.; Singer, R.H.; Segall, J. THE GREAT ESCAPE: When Cancer Cells Hijack the Genes for Chemotaxis and Motility. Annu. Rev. Cell Dev. Boil. 2005, 21, 695–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Saxton, M.J.; Jacobson, K. SINGLE-PARTICLE TRACKING:Applications to Membrane Dynamics. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 373–399. [Google Scholar] [CrossRef]
- Qian, H.; Sheetz, M.; Elson, E. Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 1991, 60, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Metzler, R.; Jeon, J.-H.; Cherstvy, A.G.; Barkai, E. Anomalous diffusion models and their properties: Non-stationarity, non-ergodicity, and ageing at the centenary of single particle tracking. Phys. Chem. Chem. Phys. 2014, 16, 24128–24164. [Google Scholar] [CrossRef] [Green Version]
- Martin, U.S.; Forstner, M.; Käs, J.A. Apparent subdiffusion inherent to single particle tracking. Biophys. J. 2002, 83, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Ruthardt, N.; Lamb, D.C.; Bräuchle, C. Single-particle Tracking as a Quantitative Microscopy-based Approach to Unravel Cell Entry Mechanisms of Viruses and Pharmaceutical Nanoparticles. Mol. Ther. 2011, 19, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Montiel, D.; Cang, H.; Yang, H. Quantitative Characterization of Changes in Dynamical Behavior for Single-Particle Tracking Studies. J. Phys. Chem. B 2006, 110, 19763–19770. [Google Scholar] [CrossRef] [PubMed]
- Dosset, P.; Rassam, P.; Fernandez, L.; Espenel, C.; Rubinstein, E.; Margeat, E.; Milhiet, P.-E. Automatic detection of diffusion modes within biological membranes using back-propagation neural network. BMC Bioinform. 2016, 17, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, R.; Manfroi, A.; Young, W. Strongly and weakly self-similar diffusion. Phys. D Nonlinear Phenom. 2001, 154, 111–137. [Google Scholar] [CrossRef]
- Sbalzarini, I.; Koumoutsakos, P. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Boil. 2005, 151, 182–195. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Corsi, F.; Mencattini, A.; Comes, M.C.; Casti, P.; di Natale, C.; Ghibelli, L.; Martinelli, E. Learning cancer-related drug efficacy exploiting consensus in clustered cell motility. IEEE Trans. Biomed. Eng. 2019. [Google Scholar] [CrossRef]
- Parlato, S.; De Ninno, A.; Molfetta, R.; Toschi, E.; Salerno, D.; Mencattini, A.; Romagnoli, G.; Fragale, A.; Roccazzello, L.; Buoncervello, M.; et al. 3D Microfluidic model for evaluating immunotherapy efficacy by tracking dendritic cell behaviour toward tumor cells. Sci. Rep. 2017, 7, 1093. [Google Scholar] [CrossRef]
- Biselli, E.; Agliari, E.; Barra, A.; Bertani, F.R.; Gerardino, A.; De Ninno, A.; Mencattini, A.; Di Giuseppe, D.; Mattei, F.; Schiavoni, G.; et al. Organs on chip approach: A tool to evaluate cancer -immune cells interactions. Sci. Rep. 2017, 7, 12737. [Google Scholar] [CrossRef]
- Davies, E.R. Machine Vision: Theory, Algorithms, Practicalities; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Gonzalez, R.C.; Woods, R.E.; Eddins, S.L. Digital Image Processing Using MATLAB; Pearson-Prentice-Hall: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Chenouard, N.; Smal, I.; De Chaumont, F.; Maška, M.; Sbalzarini, I.; Gong, Y.; Cardinale, J.; Carthel, C.; Coraluppi, S.; Winter, M.; et al. Objective comparison of particle tracking methods. Nat. Methods 2014, 11, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Szabó, B.; Szöllősi, G.J.; Gönci, B.; Jurányi, Z.; Selmeczi, D.; Vicsek, T. Phase transition in the collective migration of tissue cells: Experiment and model. Phys. Rev. E 2006, 74, 061908. [Google Scholar] [CrossRef] [Green Version]
- Yamao, M.; Naoki, H.; Ishii, S. Multi-Cellular Logistics of Collective Cell Migration. PLoS ONE 2011, 6, e27950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stichel, D.; Middleton, A.M.; Müller, B.F.; Depner, S.; Klingmüller, U.; Breuhahn, K.; Matthäus, F. An individual-based model for collective cancer cell migration explains speed dynamics and phenotype variability in response to growth factors. NPJ Syst. Boil. Appl. 2017, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda, N.; Petitjean, L.; Cochet-Escartin, O.; Grasland-Mongrain, E.; Silberzan, P.; Hakim, V. Collective Cell Motion in an Epithelial Sheet Can Be Quantitatively Described by a Stochastic Interacting Particle Model. PLoS Comput. Boil. 2013, 9, e1002944. [Google Scholar] [CrossRef] [Green Version]
- Masuzzo, P.; Van Troys, M.; Ampe, C.; Martens, L.; Information, P.E.K.F.C. Taking Aim at Moving Targets in Computational Cell Migration. Trends Cell Boil. 2016, 26, 88–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time-Varying Descriptors | |
| Tangential speed magnitude [36] | |
| Track curvature [26] | |
| Angular speed magnitude [26] | |
| Turning angle [36] | |
| Distance to track center [26] | : the distance of each track point from the corresponding track geometrical center (as the average coordinates in x and y). |
| Constant Descriptors | |
| Distance to cluster center [26] | : the average distance between the geometrical center of the entire cluster (as the average coordinates of all the track centers) and the geometrical center of the track. |
| Diffusion coefficient [25] | |
| Directional persistence [26] | : the ratio of the distance between the starting and the ending point of the track and the actual length of the track. |
| Migration speed [26] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comes, M.C.; Mencattini, A.; Di Giuseppe, D.; Filippi, J.; D’Orazio, M.; Casti, P.; Corsi, F.; Ghibelli, L.; Di Natale, C.; Martinelli, E. A Camera Sensors-Based System to Study Drug Effects on In Vitro Motility: The Case of PC-3 Prostate Cancer Cells. Sensors 2020, 20, 1531. https://doi.org/10.3390/s20051531
Comes MC, Mencattini A, Di Giuseppe D, Filippi J, D’Orazio M, Casti P, Corsi F, Ghibelli L, Di Natale C, Martinelli E. A Camera Sensors-Based System to Study Drug Effects on In Vitro Motility: The Case of PC-3 Prostate Cancer Cells. Sensors. 2020; 20(5):1531. https://doi.org/10.3390/s20051531
Chicago/Turabian StyleComes, Maria Colomba, Arianna Mencattini, Davide Di Giuseppe, Joanna Filippi, Michele D’Orazio, Paola Casti, Francesca Corsi, Lina Ghibelli, Corrado Di Natale, and Eugenio Martinelli. 2020. "A Camera Sensors-Based System to Study Drug Effects on In Vitro Motility: The Case of PC-3 Prostate Cancer Cells" Sensors 20, no. 5: 1531. https://doi.org/10.3390/s20051531