

KEAP1-Mutant Lung Cancers Weaken Anti-Tumor Immunity and Promote an M2-like Macrophage Phenotype

Abstract
:1. Introduction
2. Results
2.1. Development and Validation of KEAP1 and NRF2 Knockout Cancer Cell Lines
2.2. KEAP1 KO Enhances a Cancer-Cell Intrinsic Immunosuppressive Phenotype
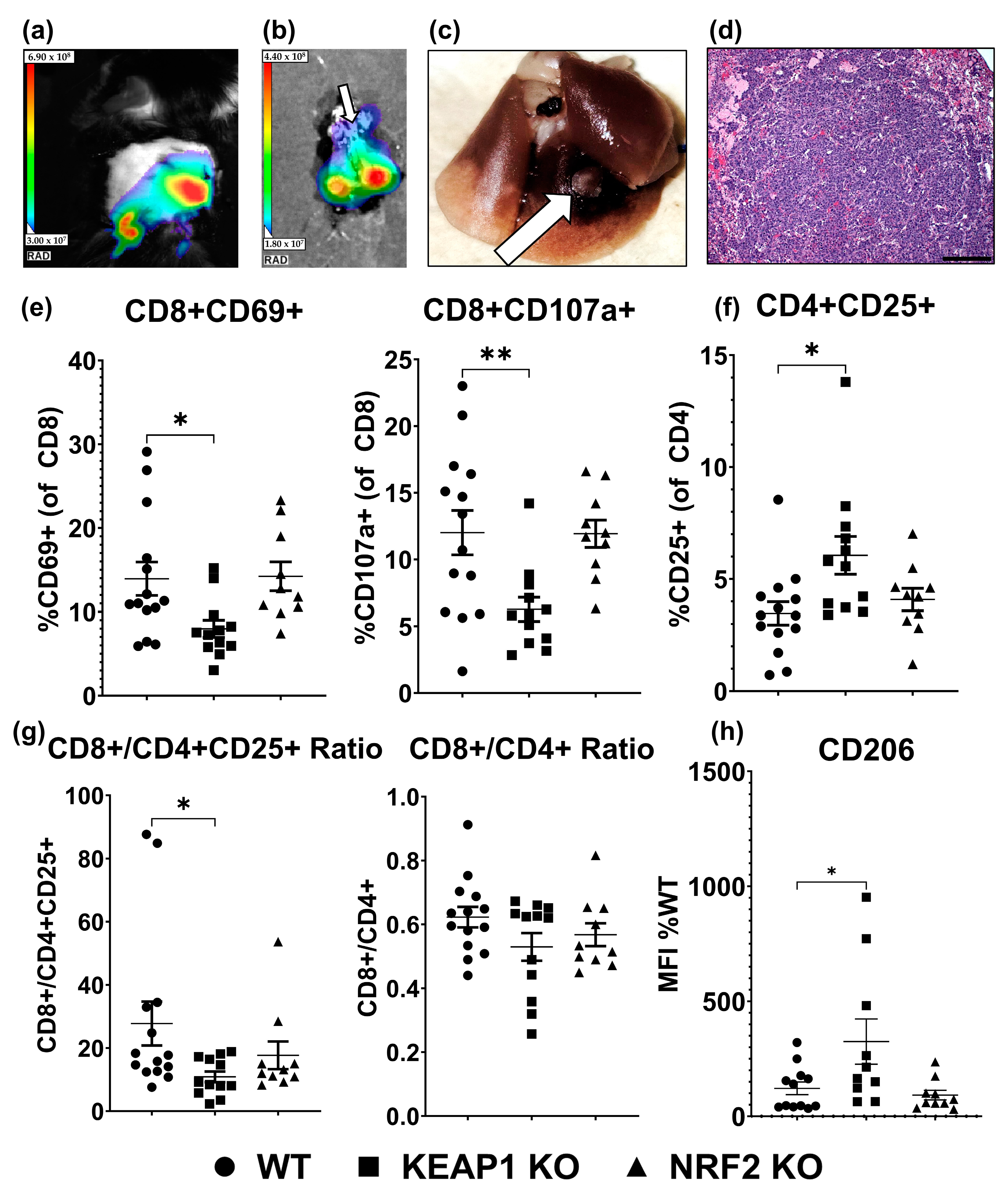
2.3. KEAP1 KO Suppresses T-Cell Function in an Orthotopic Allograft Model of Lung Cancer
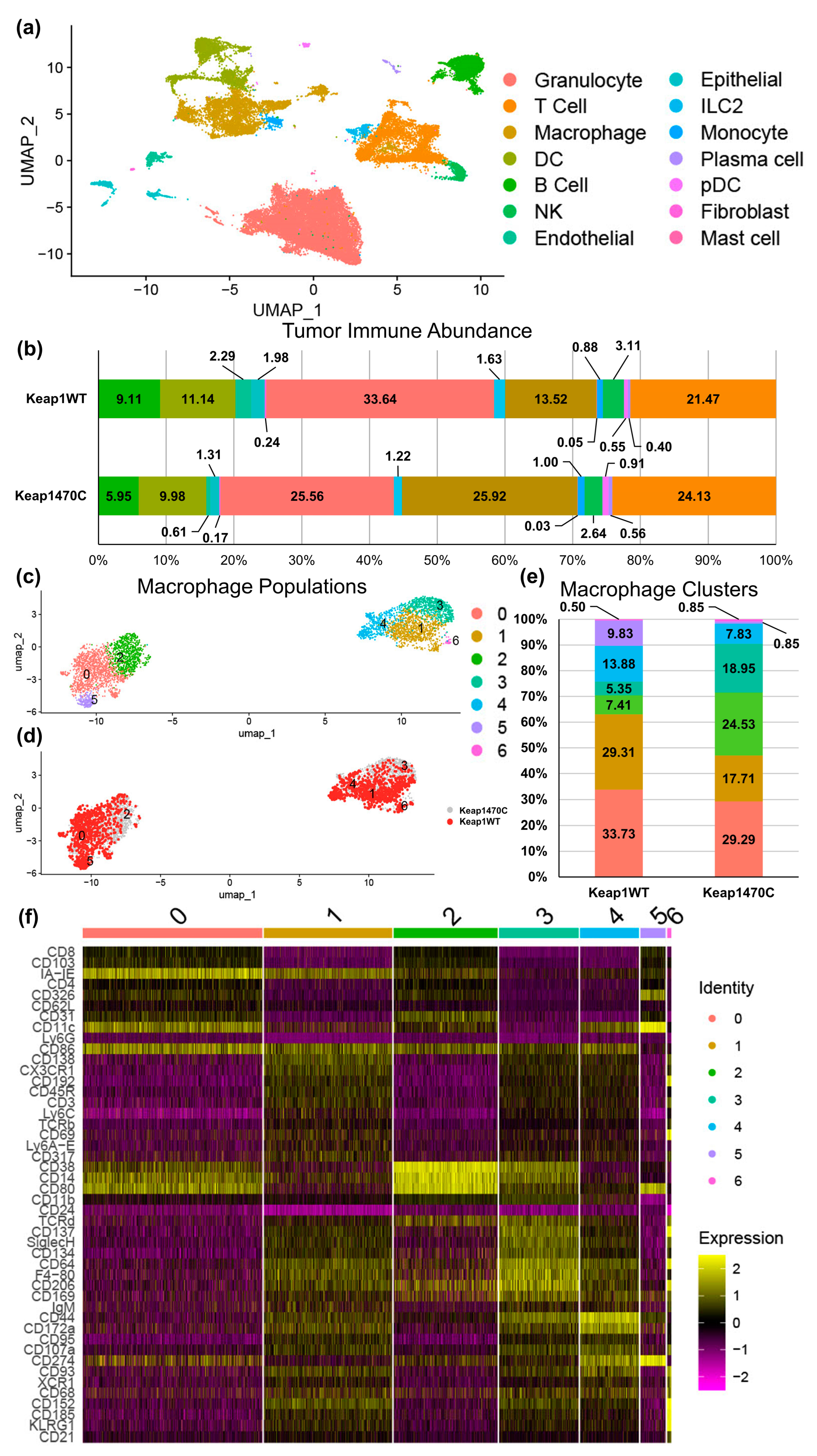
2.4. KEAP1-Mutant Lung Cancer Increases Recruitment of Macrophages
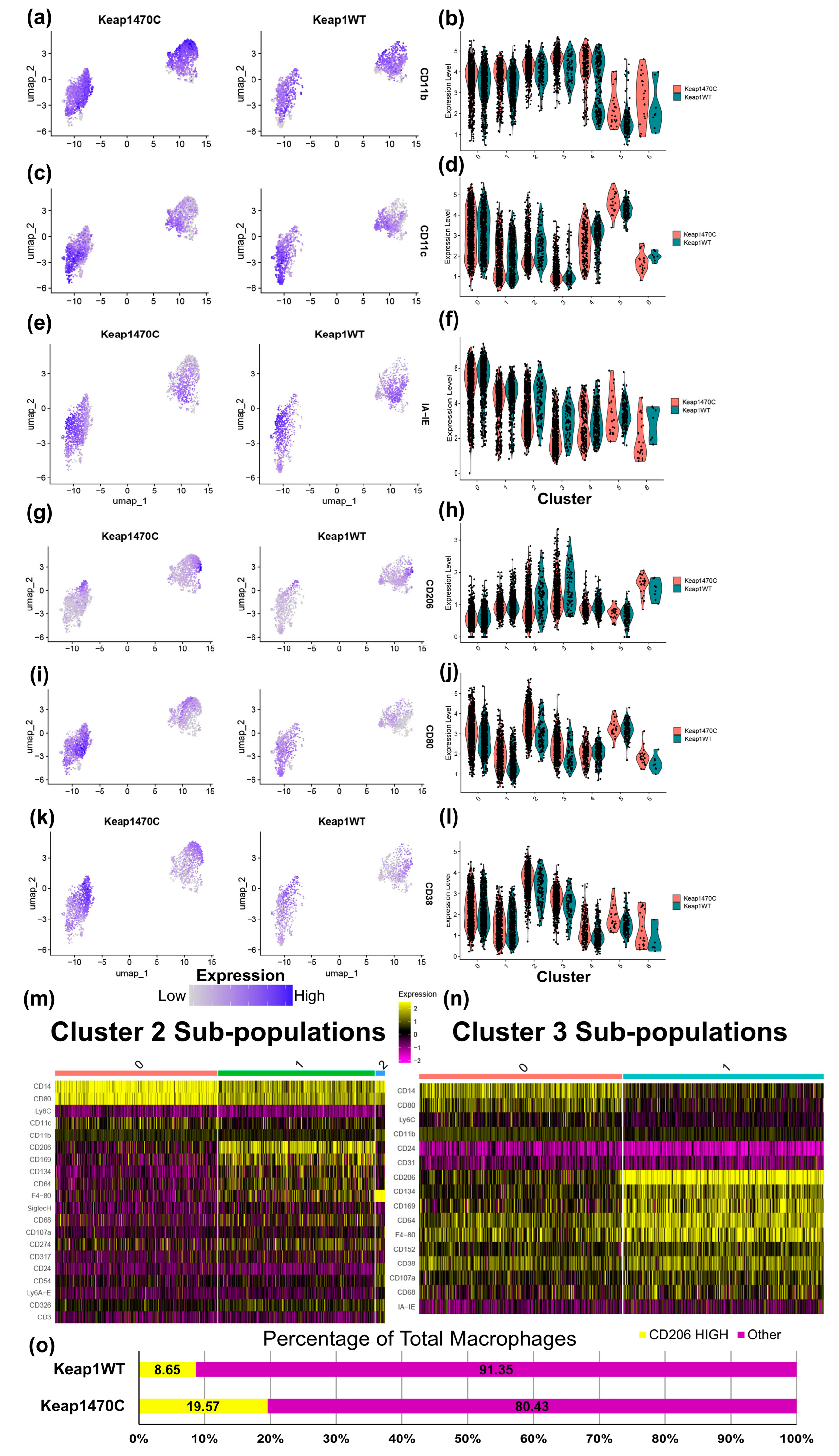
2.5. Macrophage Clusters 2 and 3 Express Markers Consistent with Infiltrating Macrophages and Poor Prognosis
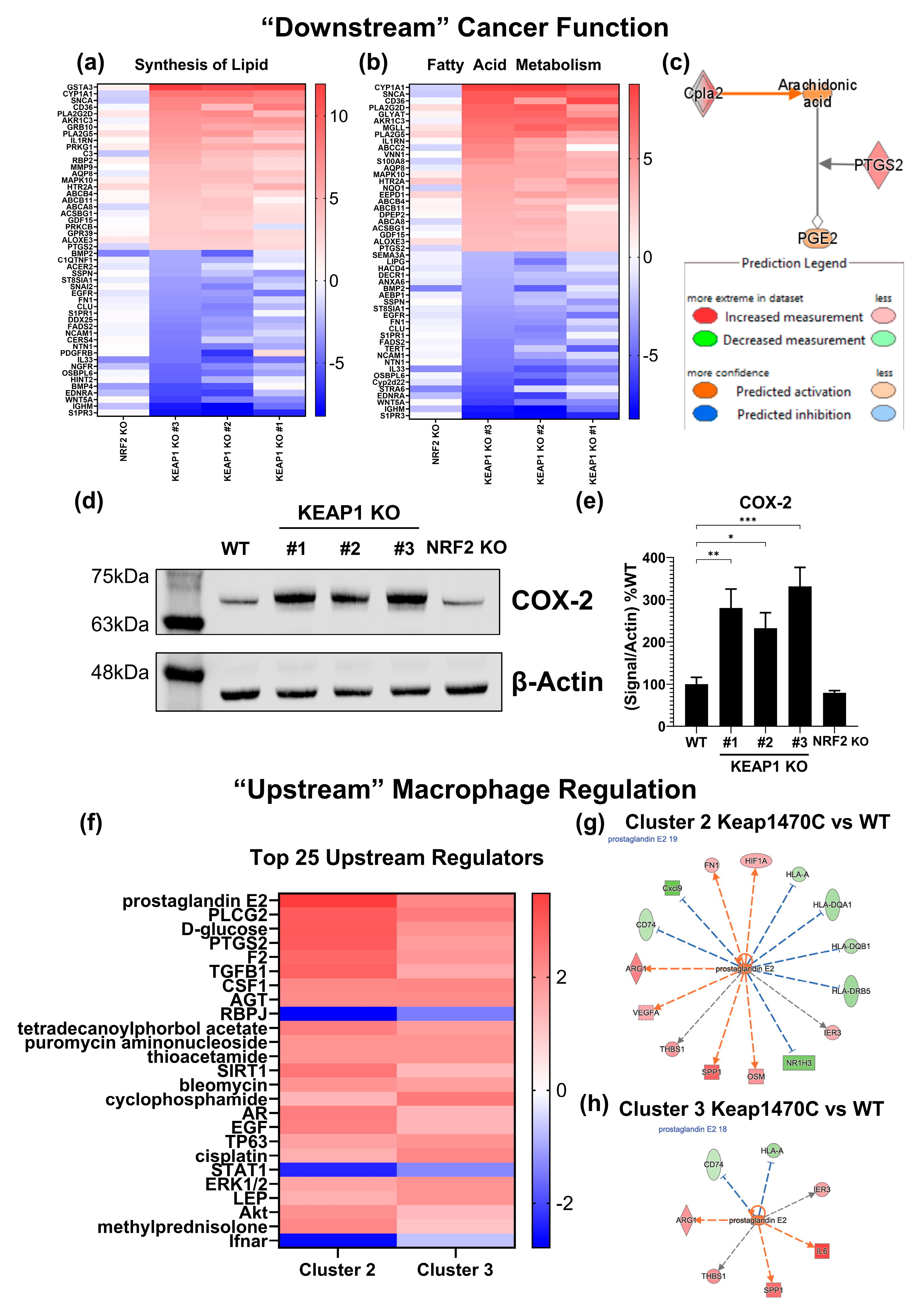
2.6. Prostaglandin Signaling May Mediate M2-like Macrophage Polarization in Keap1-Mutant Tumors
3. Discussion
4. Materials and Methods
4.1. Tissue Culture
4.2. Cell Line Creation
4.3. Western Blotting
4.4. Bulk RNA Sequencing
4.5. Protein Multiplex
4.6. RT-qPCR
4.7. Cancer Line Surface Marker Staining
4.8. Mice and Orthotopic Injection Model
4.9. Immunophenotyping
4.10. Single-Cell Analysis
4.11. QIAGEN Ingenuity Pathway Analysis Software
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients with Non–Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Chen, R.; Manochakian, R.; James, L.; Azzouqa, A.G.; Shi, H.; Zhang, Y.; Zhao, Y.; Zhou, K.; Lou, Y. Emerging Therapeutic Agents for Advanced Non-Small Cell Lung Cancer. J. Hematol. Oncol. 2020, 13, 58. [Google Scholar] [CrossRef]
- Punekar, S.R.; Shum, E.; Grello, C.M.; Lau, S.C.; Velcheti, V. Immunotherapy in Non-Small Cell Lung Cancer: Past, Present, and Future Directions. Front. Oncol. 2022, 12, 877594. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Wakabayashi, N.; Kensler, T.W. Keap1/Nrf2 Pathway in the Frontiers of Cancer and Non-Cancer Cell Metabolism. Biochem. Soc. Trans. 2015, 43, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Deblasi, J.M.; Denicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 3023. [Google Scholar] [CrossRef]
- Hu, Y.; Luo, Y.; Zheng, Y. Nrf2 Pathway and Autophagy Crosstalk: New Insights into Therapeutic Strategies for Ischemic Cerebral Vascular Diseases. Antioxidants 2022, 11, 1747. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The Emerging Role of Nrf2 in Mitochondrial Function. Free Radic. Biol. Med. 2015, 88, 179. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Ryan, D.G.; Knatko, E.V.; Casey, A.M.; Hukelmann, J.L.; Dayalan Naidu, S.; Brenes, A.J.; Ekkunagul, T.; Baker, C.; Higgins, M.; Tronci, L.; et al. Nrf2 Activation Reprograms Macrophage Intermediary Metabolism and Suppresses the Type I Interferon Response. iScience 2022, 25, 103827. [Google Scholar] [CrossRef]
- Feng, R.; Morine, Y.; Ikemoto, T.; Imura, S.; Iwahashi, S.; Saito, Y.; Shimada, M. Nrf2 Activation Drive Macrophages Polarization and Cancer Cell Epithelial-Mesenchymal Transition during Interaction. Cell Commun. Signal. 2018, 16, 54. [Google Scholar] [CrossRef]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid Modulation of IL-17 and Nrf-2 Expression Ameliorates Neuroinflammation and Promotes Remyelination in Autoimmune Encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.; Yan, W.; Xu, L.; Wang, X.; Zhao, X.; Yang, X.; Chen, G.; Ji, Y. Disruption of Nrf2 Enhances Upregulation of Nuclear Factor-KappaB Activity, Proinflammatory Cytokines, and Intercellular Adhesion Molecule-1 in the Brain after Traumatic Brain Injury. Mediators Inflamm. 2008, 2008, 725174. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-ΚB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621. [Google Scholar] [CrossRef]
- Chen, X.; Su, C.; Ren, S.; Zhou, C.; Jiang, T. Pan-Cancer Analysis of KEAP1 Mutations as Biomarkers for Immunotherapy Outcomes. Ann. Transl. Med. 2020, 8, 141. [Google Scholar] [CrossRef]
- Pillai, R.; Hayashi, M.; Zavitsanou, A.M.; Papagiannakopoulos, T. NRF2: KEAPing Tumors Protected. Cancer Discov. 2022, 12, 625. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-Occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Arbour, K.C.; Lin, J.J.; Vajdi, A.; Vokes, N.; Hong, L.; Zhang, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J. Thorac. Oncol. 2022, 17, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.C.; Zhen, Y.Y.; Niu, S.W.; Hsu, J.F.; Lee, T.H.; Chuang, H.H.; Wang, P.H.; Lee, S.C.; Lin, P.C.; Chiu, Y.W.; et al. Lung Cancer Cell-Derived Secretome Mediates Paraneoplastic Inflammation and Fibrosis in Kidney in Mice. Cancers 2020, 12, 3561. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune Evasion in Cancer: Mechanistic Basis and Therapeutic Strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Wu, G.; Xiong, R.; Sun, X.; Xu, M.; Mei, Y.; Wu, X. Tumor Immune Microenvironment Characteristics and Their Prognostic Value in Non-Small-Cell Lung Cancer. Front. Oncol. 2021, 11, 324. [Google Scholar] [CrossRef]
- Shinohara, S.; Takahashi, Y.; Komuro, H.; Matsui, T.; Sugita, Y.; Demachi-Okamura, A.; Muraoka, D.; Takahara, H.; Nakada, T.; Sakakura, N.; et al. New Evaluation of the Tumor Immune Microenvironment of Non-Small Cell Lung Cancer and Its Association with Prognosis. J. Immunother. Cancer 2022, 10, e003765. [Google Scholar] [CrossRef]
- Belluomini, L.; Dodi, A.; Caldart, A.; Kadrija, D.; Sposito, M.; Casali, M.; Sartori, G.; Ferrara, M.G.; Avancini, A.; Bria, E.; et al. A Narrative Review on Tumor Microenvironment in Oligometastatic and Oligoprogressive Non-Small Cell Lung Cancer: A Lot Remains to Be Done. Transl. Lung Cancer Res. 2021, 10, 3369. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-Tumor Genomic Biomarkers for PD-1 Checkpoint Blockade-Based Immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Zavitsanou, A.-M.; Pillai, R.; Hao, Y.; Wu, W.L.; Bartnicki, E.; Karakousi, T.; Rajalingam, S.; Herrera, A.; Karatza, A.; Rashidfarrokhi, A.; et al. KEAP1 Mutation in Lung Adenocarcinoma Promotes Immune Evasion and Immunotherapy Resistance. Cell Rep. 2023, 42, 113295. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.S.; Janik, P. Establishment of a Cloned Line of Lewis Lung Carcinoma Cells Adapted to Cell Culture. Cancer Lett. 1980, 11, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. The Diverse Functionality of NQO1 and Its Roles in Redox Control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Wang, X.; Chang, S.; Wang, T.; Wu, R.; Huang, Z.; Sun, J.; Liu, J.; Yu, Y.; Mao, Y. IL7R Is Correlated with Immune Cell Infiltration in the Tumor Microenvironment of Lung Adenocarcinoma. Front. Pharmacol. 2022, 13, 502. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in Cancer: From Biology to Therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Lo, C.-H.; Lee, S.-C.; Wu, P.-Y.; Pan, W.-Y.; Su, J.; Cheng, C.-W.; Roffler, S.R.; Chiang, B.-L.; Lee, C.-N.; Wu, C.-W.; et al. Antitumor and Antimetastatic Activity of IL-23. J. Immunol. 2003, 171, 600–607. [Google Scholar] [CrossRef]
- Kan, T.; Feldman, E.; Timaner, M.; Raviv, Z.; Shen-Orr, S.; Aronheim, A.; Shaked, Y. IL-31 Induces Antitumor Immunity in Breast Carcinoma. J. Immunother. Cancer 2020, 8, e001010. [Google Scholar] [CrossRef] [PubMed]
- Andreone, S.; Gambardella, A.R.; Mancini, J.; Loffredo, S.; Marcella, S.; La Sorsa, V.; Varricchi, G.; Schiavoni, G.; Mattei, F. Anti-Tumorigenic Activities of IL-33: A Mechanistic Insight. Front. Immunol. 2020, 11, 3072. [Google Scholar] [CrossRef]
- Deng, J.; Ma, X.; Ni, Y.; Li, X.; Xi, W.; Tian, M.; Zhang, X.; Xiang, M.; Deng, W.; Song, C.; et al. Identification of CXCL5 Expression as a Predictive Biomarker Associated with Response and Prognosis of Immunotherapy in Patients with Non-Small Cell Lung Cancer. Cancer Med. 2022, 11, 1787–1795. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, J.; Tang, D.; Zhou, L.; Chou, L.; Chou, K.-Y.; Tao, L.; Lu, L. ming Neutrophil Infiltration Mediated by CXCL5 Accumulation in the Laryngeal Squamous Cell Carcinoma Microenvironment: A Mechanism by Which Tumour Cells Escape Immune Surveillance. Clin. Immunol. 2017, 175, 34–40. [Google Scholar] [CrossRef]
- Simoncello, F.; Piperno, G.M.; Caronni, N.; Amadio, R.; Cappelletto, A.; Canarutto, G.; Piazza, S.; Bicciato, S.; Benvenuti, F. CXCL5-Mediated Accumulation of Mature Neutrophils in Lung Cancer Tissues Impairs the Differentiation Program of Anticancer CD8 T Cells and Limits the Efficacy of Checkpoint Inhibitors. Oncoimmunology 2022, 11, 2059876. [Google Scholar] [CrossRef]
- Horton, B.; Spranger, S. A Tumor Cell-Intrinsic Yin-Yang Determining Immune Evasion. Immunity 2018, 49, 11–13. [Google Scholar] [CrossRef]
- Hu, J.; Zhao, Q.; Kong, L.Y.; Wang, J.; Yan, J.; Xia, X.; Jia, Z.; Heimberger, A.B.; Li, S. Regulation of Tumor Immune Suppression and Cancer Cell Survival by CXCL1/2 Elevation in Glioblastoma Multiforme. Sci. Adv. 2021, 7, eabc2511. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-Cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune Checkpoints and Their Inhibition in Cancer and Infectious Diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef]
- Vackova, J.; Polakova, I.; Johari, S.D.; Smahel, M. CD80 Expression on Tumor Cells Alters Tumor Microenvironment and Efficacy of Cancer Immunotherapy by CTLA-4 Blockade. Cancers 2021, 13, 1935. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Inoue, H.; Mori, M.; Akiyoshi, T. The Expression of Costimulatory Molecules CD80 and CD86 in Human Carcinoma Cell Lines: Its Regulation by Interferon Gamma and Interleukin-10. Cancer Immunol. Immunother. 1996, 43, 213–219. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A Moving Target in Immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef]
- Molfetta, R.; Zitti, B.; Lecce, M.; Milito, N.D.; Stabile, H.; Fionda, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R. CD155: A Multi-Functional Molecule in Tumor Progression. Int. J. Mol. Sci. 2020, 21, 922. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Antony, P.A.; Restifo, N.P. CD4+CD25+ T Regulatory Cells, Immunotherapy of Cancer, and Interleukin-2. J. Immunother. 2005, 28, 120–128. [Google Scholar] [CrossRef]
- McNally, A.; Hill, G.R.; Sparwasser, T.; Thomas, R.; Steptoe, R.J. CD4+CD25+ Regulatory T Cells Control CD8+ T-Cell Effector Differentiation by Modulating IL-2 Homeostasis. Proc. Natl. Acad. Sci. USA 2011, 108, 7529–7534. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The Ratios of CD8+ T Cells to CD4+CD25+ FOXP3+ and FOXP3− T Cells Correlate with Poor Clinical Outcome in Human Serous Ovarian Cancer. PLoS ONE 2013, 8, 80063. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yao, G.; Zhang, Y.; Gao, J.; Yang, B.; Rao, Z.; Gao, J. M2-Polarized Tumor-Associated Macrophages Are Associated with Poor Prognoses Resulting from Accelerated Lymphangiogenesis in Lung Adenocarcinoma. Clinics 2011, 66, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of Tumor-Associated Macrophages from MHC Class IIhito MHC Class IIlowmediates Tumor Progression in Mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef]
- Gao, L.; Liu, Y.; Du, X.; Ma, S.; Ge, M.; Tang, H.; Han, C.; Zhao, X.; Liu, Y.; Shao, Y.; et al. The Intrinsic Role and Mechanism of Tumor Expressed-CD38 on Lung Adenocarcinoma Progression. Cell Death Dis. 2021, 12, 680. [Google Scholar] [CrossRef]
- Xu, K.; Ma, J.; Hall, S.R.R.; Peng, R.-W.; Yang, H.; Yao, F. Battles against Aberrant KEAP1-NRF2 Signaling in Lung Cancer: Intertwined Metabolic and Immune Networks. Theranostics 2023, 13, 704–723. [Google Scholar] [CrossRef]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct Initiating Events Underpin the Immune and Metabolic Heterogeneity of KRAS-Mutant Lung Adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef]
- Kitano, Y.; Baba, Y.; Nakagawa, S.; Miyake, K.; Iwatsuki, M.; Ishimoto, T.; Yamashita, Y.I.; Yoshida, N.; Watanabe, M.; Nakao, M.; et al. Nrf2 Promotes Oesophageal Cancer Cell Proliferation via Metabolic Reprogramming and Detoxification of Reactive Oxygen Species. J. Pathol. 2018, 244, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Yoshida, R.; Kawahara, K.; Sakata, J.; Arita, H.; Nkashima, H.; Takahashi, N.; Hirayama, M.; Nagata, M.; Hirosue, A.; et al. The Antioxidative Stress Regulator Nrf2 Potentiates Radioresistance of Oral Squamous Cell Carcinoma Accompanied with Metabolic Modulation. Lab. Investig. 2022, 102, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence InKRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Bayerl, F.; Meiser, P.; Donakonda, S.; Hirschberger, A.; Lacher, S.B.; Pedde, A.M.; Hermann, C.D.; Elewaut, A.; Knolle, M.; Ramsauer, L.; et al. Tumor-Derived Prostaglandin E2 Programs CDC1 Dysfunction to Impair Intratumoral Orchestration of Anti-Cancer T Cell Responses. Immunity 2023, 56, 1341–1358.e11. [Google Scholar] [CrossRef]
- He, X.; Stuart, J.M. Prostaglandin E2 Selectively Inhibits Human CD4+ T Cells Secreting Low Amounts of Both IL-2 and IL-4. J. Immunol. 1999, 163, 6173–6179. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kaliberov, S.; Daurkin, I.; Kaliberova, L.; Buchsbaum, D.; Vieweg, J.; Kusmartsev, S. Altered Expression of 15-Hydroxyprostaglandin Dehydrogenase in Tumor-Infiltrated CD11b Myeloid Cells: A Mechanism for Immune Evasion in Cancer. J. Immunol. 2009, 182, 7548–7557. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Xu, B.; Zhang, H.; Fang, S. Lung Adenocarcinoma Patients with KEAP1 Mutation Harboring Low Immune Cell Infiltration and Low Activity of Immune Environment. Thorac. Cancer 2021, 12, 2458. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The Necrosome Promotes Pancreatic Oncogenesis via CXCL1 and Mincle-Induced Immune Suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive Analyses of Tumor Immunity: Implications for Cancer Immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T Cell Dysfunction and Exclusion Predict Cancer Immunotherapy Response. Nat. Med. 2018, 24, 1550. [Google Scholar] [CrossRef]
- Akbari, B.; Soltantoyeh, T.; Shahosseini, Z.; Jadidi-Niaragh, F.; Hadjati, J.; Brown, C.E.; Mirzaei, H.R. PGE2-EP2/EP4 Signaling Elicits MesoCAR T Cell Immunosuppression in Pancreatic Cancer. Front. Immunol. 2023, 14, 1209572. [Google Scholar] [CrossRef]
- Shimizu, K.; Yukawa, T.; Okita, R.; Saisho, S.; Maeda, A.; Nojima, Y.; Nakata, M. Cyclooxygenase-2 Expression Is a Prognostic Biomarker for Non-Small Cell Lung Cancer Patients Treated with Adjuvant Platinum-Based Chemotherapy. World J. Surg. Oncol. 2015, 13, 21. [Google Scholar] [CrossRef]
- Conejo-Garcia, J.R. Breaking Barriers for T Cells by Targeting the EPHA2/TGF-β/COX-2 Axis in Pancreatic Cancer. J. Clin. Invest. 2019, 129, 3521–3523. [Google Scholar] [CrossRef]
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy with Immunotherapy in Cancer. Front. Oncol. 2021, 11, 637504. [Google Scholar] [CrossRef]
- Berry, A.S.F.; Amorim, C.F.; Berry, C.L.; Syrett, C.M.; English, E.D.; Beiting, D.P. An Open-Source Toolkit to Expand Bioinformatics Training in Infectious Diseases. mBio 2021, 12, e0121421. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Phipson, B.; Lee, S.; Majewski, I.J.; Alexander, W.S.; Smyth, G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Moerland, J.A.; Leal, A.S.; Lockwood, B.; Demireva, E.Y.; Xie, H.; Krieger-Burke, T.; Liby, K.T. The Triterpenoid CDDO-Methyl Ester Redirects Macrophage Polarization and Reduces Lung Tumor Burden in a Nrf2-Dependent Manner. Antioxidants 2023, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QIAGEN IPA Activation Z-Score Prediction | ||||
|---|---|---|---|---|
| NRF2 KO | KEAP1 KO #3 | KEAP1 KO #2 | KEAP1 KO #1 | |
| Xenobiotic Metabolism AHR Signaling Pathway | −2.449 | 2.309 | 2.496 | 2.53 |
| Pulmonary Fibrosis Idiopathic Signaling Pathway | −3.677 | −3.576 | −2.191 | |
| Glutathione-mediated Detoxification | −2.236 | 2.646 | 2.646 | 1.633 |
| G Beta Gamma Signaling | −3.153 | −3 | −2.646 | |
| Xenobiotic Metabolism General Signaling Pathway | −2.449 | 2.673 | 1.886 | 1.604 |
| Role of NFAT in Cardiac Hypertrophy | −3.13 | −3.128 | −2.183 | |
| CREB Signaling in Neurons | −2.562 | −3.048 | −2.722 | |
| Myelination Signaling Pathway | −2.469 | −3.111 | −2.746 | |
| Acetylcholine Receptor Signaling Pathway | −2.858 | −2.414 | −2.683 | |
| ID1 Signaling Pathway | −2.449 | −3 | −2.496 | |
| Dermatan Sulfate Biosynthesis (Late Stages) | −2.646 | −2.828 | −2.449 | |
| Chronic Myeloid Leukemia Signaling | 2 | −1.667 | −3.202 | −0.943 |
| Estrogen Receptor Signaling | −2.191 | −2.828 | −2.683 | |
| GPCR-Mediated Nutrient Sensing in Enteroendocrine Cells | −2.84 | −2.324 | −2.53 | |
| MIF-mediated Glucocorticoid Regulation | 2.646 | 2.449 | 2.449 | |
| Netrin Signaling | −2.53 | −2.714 | −2.236 | |
| Hepatic Fibrosis Signaling Pathway | −2.714 | −3.286 | −1.461 | |
| Cardiac Hypertrophy Signaling (Enhanced) | 0 | −2.48 | −2.832 | −2.137 |
| Chondroitin Sulfate Biosynthesis (Late Stages) | −2.236 | −2.646 | −2.449 | |
| WNT/Ca+ pathway | −2.53 | −2.673 | −2.121 | |
| Role of Osteoclasts in Rheumatoid Arthritis Signaling Pathway | −2.795 | −2.777 | −1.569 | |
| MIF Regulation of Innate Immunity | 2.333 | 2.121 | 2.646 | |
| Transcriptional Regulatory Network in Embryonic Stem Cells | −2.065 | −2.646 | −2.333 | |
| Factors Promoting Cardiogenesis in Vertebrates | 0 | −2.6 | −3.024 | −1.414 |
| NRF2-mediated Oxidative Stress Response | 3.153 | 1.807 | 1.732 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Occhiuto, C.J.; Liby, K.T. KEAP1-Mutant Lung Cancers Weaken Anti-Tumor Immunity and Promote an M2-like Macrophage Phenotype. Int. J. Mol. Sci. 2024, 25, 3510. https://doi.org/10.3390/ijms25063510
Occhiuto CJ, Liby KT. KEAP1-Mutant Lung Cancers Weaken Anti-Tumor Immunity and Promote an M2-like Macrophage Phenotype. International Journal of Molecular Sciences. 2024; 25(6):3510. https://doi.org/10.3390/ijms25063510
Chicago/Turabian StyleOcchiuto, Christopher J., and Karen T. Liby. 2024. "KEAP1-Mutant Lung Cancers Weaken Anti-Tumor Immunity and Promote an M2-like Macrophage Phenotype" International Journal of Molecular Sciences 25, no. 6: 3510. https://doi.org/10.3390/ijms25063510