
HFE Mutations in Neurodegenerative Disease as a Model of Hormesis

Abstract
:1. Introduction
2. Homeostatic Iron Regulator: HFE
2.1. HFE Variants and Iron Overload
2.2. Prevalence of HFE Variants
3. The Role of HFE Variants in Neurodegenerative Diseases
3.1. HFE Variants in Alzheimer’s Disease

{kind=link}
{kind=link}
| Study | Cohort Details | Prevalence of HFE Variants | Age of Onset | Other Findings |
|---|---|---|---|---|
| Yokoyama et al., 2015 [91] | 185 AD patients (met IWG-2 criteria) and 283 healthy controls (scored ≥ 26 on MMSE or 0 on CDR Scale, no report of cognitive decline in the prior year and no known disease mutation) from the US. | Higher prevalence of H63D in AD patients compared to controls (OR = 2.83; p = 0.00164). | Not measured. | After ApoE4, H63D is the variant with the second strongest association with AD. |
| Pulliam et al., 2003 [92] | 138 cognitively impaired patients (cognitive impairment and AD classification based on CERAD and NIA-RI criteria) and 67 healthy controls from the US. | Higher prevalence of H63D homozygosity and compound heterozygosity (H63D/C282Y) in ApoE4 carriers with higher cognitive impairment (p = 0.03). | Not measured. | |
| Percy et al., 2008 [93] | 54 sporadic AD patients (met NINCDS-ADRDA criteria for probable AD) and 58 healthy controls (matched for sex and age) from Canada. | Higher prevalence of bi-carriers of H63D and ApoE4 in female AD patients compared to female controls (OR = 7.13, CI = 2.07–24.6; p = 0.003). Lower prevalence of H63D in male AD patients compared to male controls (OR = 0.198, CI = 0.055–0.715; p = 0.020). | Not measured. | |
| Blázquez et al., 2007 [94] | 211 AD patients (met McKhann’s criteria) and 167 healthy controls from Spain. | Lower prevalence of H63D in AD patients compared to controls (18.0% vs. 29.9%; OR = 0.352, CI = 0.21–0.60; p < 0.05). No difference in prevalence of C282Y between AD patients and controls. This remained insignificant when stratifying by TF C2 presence (p-values not reported). | No difference between AD patients with or without H63D (p-value not reported). | |
| Lin et al., 2012 [95] | Meta analysis of 22 studies including 4365 AD patients and 8652 healthy controls genotyped C282Y and 17 studies including 2795 AD patients and 7424 healthy controls genotyped for H63D. | Lower prevalence of H63D in AD patients compared to controls (OR = 0.887, CI = 0.790–0.994; p = 0.037). No difference in prevalence of C282Y between AD patients and controls (OR = 1.039, CI = 0.914–1.181; p = 0.561). | Not measured. | |
| Correia et al., 2009 [96] | 113 AD patients (met DSM-IV and NINCDS-ADRDA criteria for probable AD) and 82 healthy controls from Portugal. | Lower prevalence of C282Y in AD patients compared to controls (1.3% vs. 5.8%; p = 0.00197). No difference in prevalence of H63D between AD patients and controls (17.2% vs. 20.3%; p = n.s.). | Lower prevalence of H63D in AD patients with late onset (17.2% in controls vs. 16.2% in AD patients with onset after 66; p = 0.0504 vs. 11.5% in patients with onset after 75; p = 0.0125). | |
| Kauwe et al., 2010 [100] | 1166 AD patients (met NINCDS-ADRDA criteria) and 1404 healthy controls (non-demented, matched for age and sex) from the US, UK and Canada. | No difference in prevalence of C282Y between AD patients and controls. Higher prevalence of bi-carriers of C282Y and TF C2 in AD patients compared to controls (SF = 2.4, CI = 1.38–4.19; p = 0.002). | Not measured. | |
| Robson et al., 2004 [101] | 191 AD patients (met CERAD or NINCDS-ADRDA criteria for definite or probable AD), 69 MCI patients (defined by Petersen et al., 1999 [102]) and 269 healthy controls (without cognitive impairment and scored >80 on CAMCOG) from the UK. | Higher prevalence of bi-carriers of TF C2 and C282Y in female AD patients compared to female controls (OR = 7.0, CI = 1.5–33; p = 0.006). No difference in prevalence of H63D or C282Y between AD patients, MCI patients, and controls (H63D: 27.8% vs. 24.6% vs. 27.9%; p = n.s. C282Y: 15.7% vs. 24.6% vs. 11.9%; p = n.s.) | No difference between AD patients with or without H63D, C282Y, TF C2, or any combination of these variants. | |
| Alizadeh et al., 2009 [99] | 268 AD patients (met NINCDS-ADRDA criteria) and 1970 healthy controls (no history of neurological disease) from The Netherlands. | No difference in prevalence of H63D or C282Y between AD patients and controls (p-values not reported). | Average 5.5 years earlier onset in men with ApoE4 and H63D (73.2 ± 2.1 vs. 78.7 ± 1.6 years; p = 0.05). | |
| Sampietro et al., 2001 [43] | 107 sporadic, late-onset AD patients (met NINCDS-ADRDA criteria) and 99 healthy controls (matched for age) from Italy. | No difference in prevalence of H63D between AD patients and controls (11% vs. 14%; p = 0.46). No difference in prevalence of C282Y between AD patients and controls (2% vs. 2%; p = 1.00). | Higher prevalence of H63D in AD patients with earlier onset (22% in patients with onset between 60–70 vs. 12% in patients with onset between 70–80 vs. 4% in patients with onset after 80; p = 0.004). | |
| Lehmann et al., 2012 [97] | 1757 AD patients (met CERAD or NINCDS-ADRDA criteria for definite or probable AD), and 6294 healthy controls from multiple Caucasian cohorts. | No difference in prevalence of H63D between AD patients and controls (OR = 1.0, CI = 0.8–1.1; p = 0.6). No difference in prevalence of C282Y between AD patients and controls (OR = 1.0, CI = 0.8–1.2; p = 0.8). | Lower prevalence of H63D in AD patients with onset after 80 years (p = 0.02). | |
| Combarros et al., 2003 [98] | 328 sporadic AD patients (met NINCDS-ADRDA criteria for probable AD). | Not measured. | No difference between AD patients with or without H63D (p-value not reported). Earlier onset in AD patients with H63D who are homozygous for ApoE4 (5.1 years earlier than ApoE4 heterozygotes; p = 0.014, and 6.1 years earlier than ApoE4 non-carriers; p = 0.019). | |
| Berlin et al., 2004 [44] | 213 sporadic AD patients (met NINCDS-ADRDA criteria for probable AD), 106 MCI patients (scored 0.5 on the CDR Scale) and 63 healthy controls from Canada. | No difference in prevalence of H63D between AD patients, MCI patients, and controls (33% vs. 26% vs. 34%; p = 0.43). No difference in prevalence of C282Y between AD patients, MCI patients and controls (5% vs. 6% vs. 10%; p = 0.33). | No difference between AD patients with or without H63D (p = 0.72). No difference between AD patients with or without C282Y (p = 0.93). | The presence of H63D or C282Y did not affect patients’ neuropsychological scores. No significant associations were found between ApoE4 and either H63D or C282Y. |
| Avila-gomez et al., 2008 [103] | 105 early-onset AD patients (containing the E280A variant of PSEN-1) and 220 healthy controls (non-demented and unrelated to study patients) from Colombia. | No difference in prevalence of H63D between AD patients and controls (32.4% vs. 29.57%; p = 0.8061). | No difference between AD patients with or without H63D (44.0 ± 5.1 vs. 44.42 ± 5.2 years; p = 0.7531). | |
| Candore et al., 2003 [104] | 123 AD patients and 152 healthy controls (non-demented, unrelated to study patients) from Italy. | No difference in prevalence of H63D between AD patients and controls (13.8% vs. 11.2%; p-value not reported), remained insignificant when stratifying by ApoE status. | No difference between AD patients with or without H63D (67.79 ± 9.14 vs. 67.93 ± 2.54 years; p-value not reported). | C282Y and S65C were analyzed, but both had presence too low to analyze. |
| Corder and Beaumont 2007 [105] | 90 possible AD patients, 80 probable AD patients, 145 definite AD patients (met NINCDS-ADRDA criteria for probable or possible AD or met CERAD criteria for definite AD; stratified by diagnosis) and 260 healthy controls (without cognitive dysfunction and scored >80 on CAMCOG) from the UK. | No difference in prevalence of H63D or C282Y among groups (H = 0.13 and 0.04, respectively). | Not measured. | |
| Giambattistelli et al., 2012 [106] | 160 AD patients (met NINCDS-ADRDA criteria and scored ≤ 25 on MMSE) and 79 healthy controls (no evidence of conditions known to affect mental metabolism) from Italy. | No difference in prevalence of H63D between AD patients and controls (28.1% vs. 25.3%; p = 0.38). | No difference between AD patients with or without H63D (data not reported). | No difference in disease duration between AD patients with or without H63D (data not reported). |
| Lleo et al., 2002 [107] | 108 AD patients (met NINCDS-ADRDA criteria for probable AD) and 110 healthy controls (no medical illness or cognitive impairment, unrelated to study patients) from Spain. | No difference in prevalence of H63D between AD patients and controls (42.6% vs. 34.5%; p-value not reported). No difference in prevalence of C282Y between AD patients and controls (3.7% vs. 3.6%; p-value not reported). Neither became significant when stratifying ApoE status or TF C2 presence (p-values not reported). | Not measured. | |
| Mariani et al., 2013 [108] | 139 AD patients (met NINCDS-ADRDA criteria for probable AD and scored ≤25 on MMSE), 78 PD patients (met UK PDS Brain Bank criteria), 27 MCI patients (see paper for inclusion criteria) and 139 healthy controls from Italy. | No difference in prevalence of H63D between AD patients and controls (28.3% vs. 28.8%; p = 0.4). No difference in prevalence of C282Y between AD patients and controls (0% vs. 1.4%; p = 0.1). | Not measured. | |
| Guerreiro et al., 2006 [48] | 130 AD patients (met DSM-IV and NINCDS-ADRDA criteria, had onset ≥ 65 and no family history of AD), 132 PD patients (met UK PDS Brain Bank criteria), 55 MCI patients (met criteria defined by Petersen et al., 1999 [102]), and 115 healthy controls (matched for age) from Portugal. | No difference in prevalence of H63D between AD patients and controls (33.6% vs. 35.6%; p = 0.76). No difference in prevalence of C282Y between AD patients and controls (4.6% vs. 4.3%; p = 0.92). | No difference between AD patients with or without either H63D (p = 0.93) or C282Y (p = 0.08). | |
| Vance et al., 2020 [109] | 12,532 AD patients and 13,134 controls from the Alzheimer’s Disease Genetics Consortium (ADGC). | No difference in prevalence of C282Y between AD patients and controls (p = 0.40). This remained insignificant when stratifying by TF C2 presence (p = 0.23). | Not measured. | No detection of epistasis between TF C2 and C282Y (SF = 0.94; p = 0.48). |
3.2. HFE Variants in Parkinson’s Disease
| Study | Cohort Details | Prevalence of HFE Variants | Other Findings |
|---|---|---|---|
| Greco et al., 2011 [47] | 181 sporadic PD patients (met UK PDS Brain Bank criteria) and 180 healthy controls from Italy. | Higher allele frequency of H63D in PD patients compared to controls (17.4% vs. 11.7%; p = 0.029). No difference in allele frequency of C282Y between PD patients and controls (0.8% vs. 0.8%; p = 0.995). | |
| Xia et al., 2015 [52] | Meta analysis of 8 studies including 1631 AD patients and 4548 healthy controls genotyped for C282Y and 7 studies including 1192 AD patients and 4065 healthy controls genotyped for H63D. | Lower prevalence of C282Y in PD patients compared to controls (11.47% vs. 12.03%; OR = 0.22, CI = 0.09–0.57; p = 0.002). No difference in prevalence of H63D between PD patients and controls (26.09% vs. 26.32%; OR = 0.99, CI = 0.84–1.17; p = 0.925). | |
| Buchanan et al., 2002 [112] | 438 PD patients and 485 healthy controls (non-demented with no related illnesses; matched for age and sex) from Australia. | Lower prevalence of C282Y in PD patients compared to controls (10.7% vs. 16.5%; OR = 0.56, CI = 0.33–0.94; p = 0.027). | |
| Guerreiro et al., 2006 [48] | 130 AD patients (met DSM-IV and NINCDS-ADRDA criteria, had onset ≥ 65 and no family history of AD), 132 PD patients (met UK PDS Brain Bank criteria), 55 MCI patients (met criteria defined by Petersen et al., 1999 [102]), and 115 healthy controls (matched for age) from Portugal. | Higher prevalence of C282Y in PD patients compared to controls (13.6% vs. 4.3%; p = 0.01). No difference in prevalence of H63D between PD patients and controls (32.6% vs. 35.6%; p = 0.47). | No difference in age of onset between PD patients with or without H63D (p = 0.98) or C282Y (p = 0.76). |
| Dekker et al., 2003 [51] | 197 PD patients and 72 non-PD PS patients (from two cohorts; diagnosed according to the EUROPARKIN protocol) and 7983 healthy controls (from the Rotterdam study) from The Netherlands. | Higher prevalence of C282Y homozygosity in one cohort of PD patients compared to controls (1.5% vs. 0.3%; p = 0.03). No difference in prevalence of H63D between PD patients and controls (p-value not reported). | Higher allele frequency of C282Y in non-PD PS patients compared to controls (13.7% and 17.5% in PS cohorts vs. 6.1% in controls; p = 0.005 and p = 0.002, respectively). |
| Aamodt et al., 2007 [111] | 388 PD patients (met criteria from Gelb et al., 1999 [113]), and 505 healthy controls (from Distante et al., 1999 [114]) from Norway. | No difference in prevalence of H63D between PD patients and controls (17.3% vs. 19.4%; p-value not reported). No difference in prevalence of C282Y between PD patients and controls (13.4% vs. 13.1%; p-value not reported). | |
| Borie et al., 2002 [46] | 216 PD patients (demonstrated at least two of these signs: rigidity, bradykinesia, resting tremor, asymmetric onset; had improvement with levodopa treatment and absence of exclusion criteria), and 193 healthy controls (matched for age and sex) from France. | No difference in prevalence of H63D between PD patients and controls (36.3% vs. 33.9%; p = 1.000). No difference in prevalence of C282Y between PD patients and controls (7.0% vs. 8.8%; p = 0.7506). | |
| Akbas et al., 2006 [49] | 278 idiopathic PD patients (met UK PDS Brain Bank criteria) and 280 healthy controls (no extrapyramidal disorders, matched for ethnicity) from Germany. | No difference in prevalence of H63D between PD patients and controls (16.0% vs. 14.1%; p-value not reported). No difference in prevalence of C282Y between PD patients and controls (4.7% vs. 5.8%; p-value not reported). | |
| Biasiotto et al., 2008 [50] | 475 PD patients (met UK PDS Brain Bank criteria and demonstrated bradykinesia and at least one of these signs: resting tremor, rigidity and postural instability, positive response to dopaminergic therapy and absence of atypical features or other causes of parkinsonism) and 2351 healthy controls from Italy. | No difference in prevalence of H63D between PD patients and controls (14.53% vs. 13.3%; p = 0.31). No difference in prevalence of C282Y between PD patients and controls (1.7% vs. 1.6%; p = 0.88). | No difference in age of onset between PD patients with or without H63D (data not reported). |
| Mariani et al., 2013 [108] | 139 AD patients (met NINCDS-ADRDA criteria for probable AD and scored ≤25 on MMSE), 78 PD patients (met UK PDS Brain Bank criteria), 27 MCI patients (see paper for inclusion criteria) and 139 healthy controls from Italy. | No difference in prevalence of H63D between PD patients and controls (31.2% vs. 28.8%; p = 0.5). No difference in prevalence of C282Y between PD patients and controls (4.1% vs. 1.4%; p = 0.2). | |
| Halling et al., 2008 [45] | 79 idiopathic PD patients (demonstrated 2 of these signs: tremor, bradykinesia, rigidity) and 153 healthy controls (matched for age) from the Faroe Islands. | No difference in prevalence of H63D between PD patients and controls (29% vs. 25%; p = 0.60). No difference in prevalence of C282Y between PD patients and controls (13% vs. 14%; p = 0.50). | |
| Yi et al., 2023 [115] | Meta-analysis of 9 studies where HFE variants were genotyped containing 1801 PD patients and 4796 healthy controls. | No difference in prevalence of H63D between PD patients and controls (OR = 1.02, CI = 0.87–1.21; p-value not reported). No difference in prevalence of C282Y between PD patients and controls (OR = 0.88, CI = 0.72–1.08; p-value not reported). | |
| Saini et al., 2021 [116] | 16,318 PD patients and 18,717 healthy controls from two cohorts (IPDGC and AMP-PD). | No difference in prevalence of H63D between PD patients and controls (17% vs. 16%; OR = 1.02, CI = 0.97–1.07; p = 0.53). No difference in prevalence of C282Y between PD patients and controls (5.2% vs. 5.9%; OR = 0.98, CI = 0.91–1.07; p = 0.69). |
3.3. HFE Variants in ALS
| Study | Cohort Details | Prevalence of HFE Variants | Age of Onset | Other Findings |
|---|---|---|---|---|
| Sutedja et al., 2007 [55] | 289 SALS patients (met El Escorial criteria for definite, probable, or possible ALS) and 5886 healthy controls from The Netherlands. | Higher prevalence of H63D homozygosity in ALS patients compared to controls (4.5% vs. 2.1%; OR = 2.2, CI = 1.1–4.1; p = 0.02). No difference in prevalence of H63D between ALS patients and controls (27.8% vs. 26.7%; OR = 1.1, CI = 0.8–1.4; p = 0.68). Trend toward lower prevalence of C282Y heterozygosity in ALS patients compared to controls (6.3% vs. 9.6%; OR = 0.6, CI = 0.4–1.0; p = 0.06). | Later onset in ALS patients with H63D compared to those without (62.9 vs. 58 years; p < 0.001). No difference between ALS patients with or without C282Y (56.6 vs. 59.6 years; p = 0.20). | No difference in disease duration between patients with or without H63D (2.6 vs. 3.1 years; p = 0.60). No difference in disease duration between patients with or without C282Y (3.1 vs. 3.0 years; p = 0.9). No difference in site of onset between ALS patients with or without either HFE variant (data not reported). |
| Goodall et al., 2005 [54] | 166 SALS patients (met El Escorial criteria for definite or probable ALS) and 192 healthy controls (unrelated to study patients) from the UK and 213 SALS patients and 208 healthy controls from Ireland. | Higher prevalence of H63D in ALS patients compared to controls (34.3% vs. 22.0%; p < 0.001). No difference in prevalence of C282Y between ALS patients and controls (18.2% vs. 19.0%; p = 0.783). | No difference between ALS patients with or without H63D (56.14 ± 13.04 vs. 57.81 ± 12.18 years; p = 0.249). | No difference in site of onset between ALS patients with or without H63D (30.37% vs. 26.12% bulbar; p = 0.409). |
| He et al., 2011 [117] | 195 SALS patients (met El Escorial criteria for definite or probable ALS) and 405 healthy controls (unrelated to study patients; matched for age, sex, and ethnicity) from China. | Higher prevalence of H63D in ALS patients compared to controls (10.26% vs. 3.21%; OR = 3.45, CI = 1.68–7.08; p < 0.001) | No difference between ALS patients with or without H63D (data not reported). | No difference in site of onset between ALS patients with or without H63D (data not reported). |
| Restagno et al., 2007 [56] | 149 SALS patients (met El Escorial criteria for definite or probable ALS) and 168 healthy controls (matched for age, sex, and ethnicity) from Italy. | Higher prevalence of H63D in ALS patients compared to controls (28.8% vs. 14.8%; p = 0.004). | No difference between ALS patients with or without H63D (63.4 ± 9.3 vs. 60.2 ± 11.9 years; p = 0.002). | No difference in disease duration between ALS patients with or without H63D (783 vs. 993 days; p-value not reported). No difference in site of onset between ALS patients with or without H63D (23.26% vs. 24.53% bulbar; p-value not reported). |
| Wang et al., 2004 [58] | 121 SALS patients (met El Escorial criteria for definite or probable ALS) and 133 healthy controls (normal neurological examination) from the US. | Higher prevalence of H63D in ALS patients compared to controls (29.75% vs. 14.29%; p = 0.0043) | No difference between ALS patients with or without either HFE variant (58.69 vs. 58.26 years; p = 0.8797). | No difference in disease duration between ALS patients with or without H63D (37.30 vs. 34.67 months; p = 0.6558). |
| Li et al., 2014 [62] | Meta-analysis of 6 cohorts containing 1692 ALS patients and 8359 healthy controls genotyped for C282Y and 14 cohorts containing 5849 ALS patients and 13,710 healthy controls genotyped for H63D. | Lower prevalence of C282Y in ALS patients compared to controls (8.98% vs. 10.89%; OR = 0.75, CI = 0.61–0.92; p = 0.006). Trend toward higher prevalence of H63D in ALS patients compared to controls (26.62% vs. 26.37%; OR = 1.14, CI = 0.98–1.33; p = 0.086). | Not measured. | |
| Praline et al., 2012 [61] | 824 SALS patients (met El Escorial criteria for definite or probable ALS) and 447 healthy controls (matched for age, sex, and ethnicity) from France. | Lower prevalence of C282Y in ALS patients compared to controls (6.3% vs. 11.6%; OR = 1.95, CI = 1.3–2.92; p = 0.001). No difference in H63D prevalence between ALS patients and controls (27.0% vs. 30.2%; p = 0.29). | Later onset in ALS patients with H63D compared to without (61.3 ± 11.8 vs. 59.4 ± 12.9 years; p = 0.05). No difference between ALS patients with or without C282Y (60.7 ± 11.2 vs. 59.9 ± 12.8 years; p = 0.66). | No difference in disease duration between ALS patients with or without C282Y (p = 0.73). No difference in disease duration between ALS patients with or without H63D (p = 0.25). No difference in site of onset between ALS patients with or without either variant (H63D: 31.1% vs. 28.4% bulbar; p = 0.65, C282Y: 34.0% vs. 28.5% bulbar; p = 0.42). No difference in frequency of either variant between ALS patients with or without ApoE4 (data not reported). |
| Yen et al., 2004 [59] | 51 ALS patients (met El Escorial criteria for definite or probable ALS) and 47 healthy controls from the US. | No difference in prevalence of H63D between ALS patients and controls (13.7% vs. 11.7%; p = 0.831). No difference in prevalence of C282Y between ALS patients and controls (2.9% vs. 2.1%; p = 1.000). | No difference between ALS patients with or without either HFE variant (46.5 ± 12.8 vs. 49.8 ± 13.4 years; p = 0.425). | No difference in rate of progression (change of Appel score/months) in ALS patients with or without either HFE variant (1.52 ± 1.55 vs. 2.00 ± 2.43; p = 0.547). |
| Chiò et al., 2015 [57] | 1119 ALS patients (definite, probable, probable-laboratory supported, and possible) and 1302 healthy controls (matched for age, sex, and ethnicity) from Italy and 232 ALS patients and 121 healthy controls from Sardinia (same criteria). | No difference in prevalence of H63D between Italian ALS patients and controls (28.2% vs. 27.2%; p-value not reported) or between Sardinian ALS patients and controls (33.6% vs. 34.7%; p-value not reported). | No difference between Italian ALS patients with homozygous, heterozygous, or without H63D (62.5 ± 11.2 vs. 62.2 ± 11.2 vs. 62.3 ± 11.2 years; p = 0.92) or between Sardinian ALS patients with homozygous, heterozygous, or without H63D (65.4 ± 10.3 vs. 60.6 ± 10.5 vs. 60.2 ± 12.8 years; p = 0.78). | No difference between Italian ALS patients with or without H63D (3.4 vs. 3.0 years; p = n.s.) or between Sardinian ALS patients with or without H63D (3.5 vs. 4.7 years; p = n.s.). Longer disease duration for Italian ALS patients carrying SOD1 mutations with H63D compared to without (15.3 vs. 2.1 years; p = 0.04). No difference in site of onset between ALS patients with or without H63D (26.3% vs. 26.9% bulbar; p = 0.91). |
| van Rheenen et al., 2013 [60] | 3962 ALS patients and 5072 healthy controls (without any neuromuscular disease and unrelated to study patients) from The Netherlands, Belgium, Germany, Ireland, UK, Sweden, and Switzerland. | No difference in prevalence of H63D between ALS patients and controls (26% vs. 27%; p = 0.99). | No difference between ALS patients with or without H63D (homozygous p = 0.68, heterozygous p = 0.62). | No difference in disease duration between ALS patients with or without H63D (homozygous p = 0.51, heterozygous p = 0.26). This study also completed a meta-analysis of 6 previous studies, and their findings were consistent with their own study after adding these data. |
| Canosa et al., 2023 [118] | 30 ALS patients from France and 153 ALS patients from Italy; all with SOD1 mutations. | Not measured. | No difference between ALS patients with or without H63D (p = 0.904). | Longer survival in ALS patients with H63D compared to those without (13.58 vs. 6.09 years; p = 0.034). |
| Su et al., 2013 [63] | 38 SALS patients (met El Escorial criteria for definite, probable, or possible ALS) from the US (15 H63D homozygotes, 1 H63D heterozygote, and 22 WT HFE). | Not measured. | No difference between ALS patients with or without H63D (56.5 vs. 56.7 years; p = 0.973). | Average 28.1 months longer disease duration in ALS patients with H63D compared to without (75.3 ± 12.7 vs. 47.2 ± 11.0 months; p = 0.017). No difference in site of onset between ALS patients with or without H63D (12.5% vs. 13.6% bulbar; p = 1.000) |
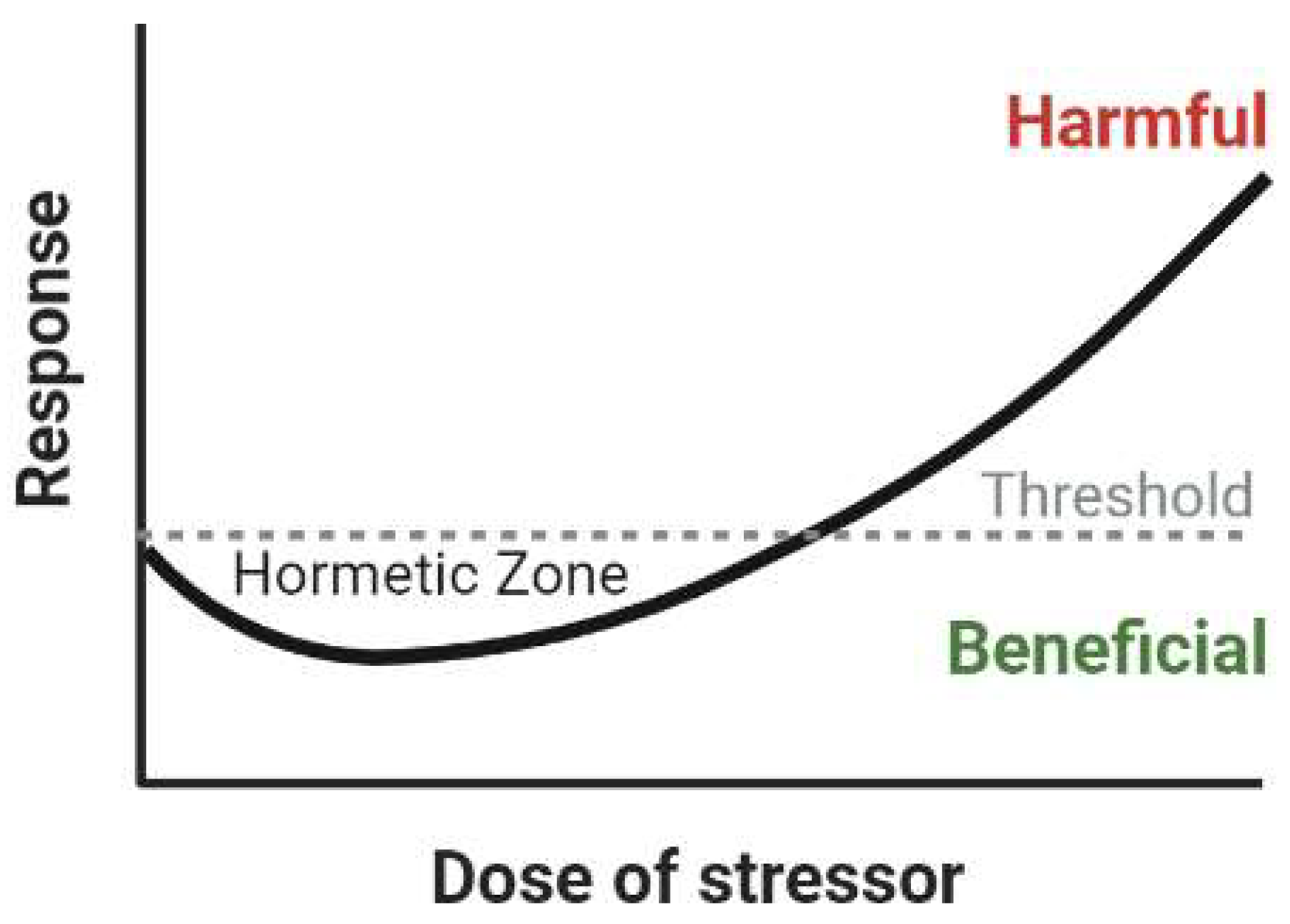
3.4. Overall Effects of HFE Variants in Neurodegenerative Disease: A Possible Mechanism of Hormesis
4. Animal and Cell Models of HFE Variants
4.1. Rodent Models
4.2. Primary Cell Culture Models
4.3. Cell Line Models
5. Mechanisms of Hormesis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nandar, W.; Connor, J.R. HFE Gene Variants Affect Iron in the Brain. J. Nutr. 2011, 141, 729S–739S. [Google Scholar] [CrossRef]
- Chiou, B.; Neal, E.H.; Bowman, A.B.; Lippmann, E.S.; Simpson, I.A.; Connor, J.R. Endothelial Cells Are Critical Regulators of Iron Transport in a Model of the Human Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2019, 39, 2117–2131. [Google Scholar] [CrossRef]
- Levenson, C.W.; Tassabehji, N.M. Iron and Ageing: An Introduction to Iron Regulatory Mechanisms. Ageing Res. Rev. 2004, 3, 251–263. [Google Scholar] [CrossRef]
- Bartzokis, G.; Cummings, J.L.; Markham, C.H.; Marmarelis, P.Z.; Treciokas, L.J.; Tishler, T.A.; Marder, S.R.; Mintz, J. MRI Evaluation of Brain Iron in Earlier- and Later-Onset Parkinson’s Disease and Normal Subjects. Magn. Reson. Imaging 1999, 17, 213–222. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra during Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef]
- Connor, J.R.; Menzies, S.L.; Martin, S.M.S.; Mufson, E.J. Cellular Distribution of Transferrin, Ferritin, and Iron in Normal and Aged Human Brains. J. Neurosci. Res. 1990, 27, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.M.; Connor, J.R. Iron, the Substantia Nigra and Related Neurological Disorders. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, B.; Sourander, P. The Effect of Age on the Non-Haemin Iron in the Human Brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Bellei, C.; Giannelli, S.; Terreni, M.R.; Gallorini, M.; Rizzio, E.; Pezzoli, G.; Albertini, A.; Zecca, L. Neuromelanin and Iron in Human Locus Coeruleus and Substantia Nigra during Aging: Consequences for Neuronal Vulnerability. J. Neural Transm. 2006, 113, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Grubić Kezele, T.; Ćurko-Cofek, B. Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients 2020, 12, 2601. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Orii, K.O.; Fleming, R.E.; Holden, C.C.; Waheed, A.; Britton, R.S.; Gutierrez, M.A.; Velez-Castrillon, S.; Bacon, B.R.; Sly, W.S. Contribution of the H63D Mutation in HFE to Murine Hereditary Hemochromatosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15788–15793. [Google Scholar] [CrossRef]
- Cornett, C.R.; Markesbery, W.R.; Ehmann, W.D. Imbalances of Trace Elements Related to Oxidative Damage in Alzheimer’s Disease Brain. Neurotoxicology 1998, 19, 339–345. [Google Scholar]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Kirsch, W.M. Iron, Zinc and Copper in the Alzheimer’s Disease Brain: A Quantitative Meta-Analysis. Some Insight on the Influence of Citation Bias on Scientific Opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.R.; Sayre, L.M.; Perry, G. Iron Accumulation in Alzheimer Disease Is a Source of Redox-Generated Free Radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Ding, B.; Chen, K.-M.; Ling, H.-W.; Sun, F.; Li, X.; Wan, T.; Chai, W.-M.; Zhang, H.; Zhan, Y.; Guan, Y.-J. Correlation of Iron in the Hippocampus with MMSE in Patients with Alzheimer’s Disease. J. Magn. Reson. Imaging 2009, 29, 793–798. [Google Scholar] [CrossRef]
- Bartzokis, G.; Tishler, T.A. MRI Evaluation of Basal Ganglia Ferritin Iron and Neurotoxicity in Alzheimer’s and Huntingon’s Disease. Cell. Mol. Biol. 2000, 46, 821–833. [Google Scholar]
- Sofic, E.; Riederer, P.; Heinsen, H.; Beckmann, H.; Reynolds, G.P.; Hebenstreit, G.; Youdim, M.B.H. Increased Iron (III) and Total Iron Content in Post Mortem Substantia Nigra of Parkinsonian Brain. J. Neural Transm. 1988, 74, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased Nigral Iron Content and Alterations in Other Metal Ions Occurring in Brain in Parkinson’s Disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Dirr, A.; Goetz, M.; Sofic, E.; Jellinger, K.; Youdim, M.B. Distribution of Iron in Different Brain Regions and Subcellular Compartments in Parkinson’s Disease. Ann. Neurol. 1992, 32, S101–S104. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Dobson, B.R.; Jones, G.R.; Clarke, D.T. Iron in the Basal Ganglia in Parkinson’s Disease. An in Vitro Study Using Extended X-Ray Absorption Fine Structure and Cryo-Electron Microscopy. Brain J. Neurol. 1999, 122 Pt 4, 667–673. [Google Scholar] [CrossRef]
- Gorell, J.M.; Ordidge, R.J.; Brown, G.G.; Deniau, J.-C.; Buderer, N.M.; Helpern, J.A. Increased Iron-related MRI Contrast in the Substantia Nigra in Parkinson’s Disease. Neurology 1995, 45, 1138–1143. [Google Scholar] [CrossRef]
- Ryvlin, P.; Broussolle, E.; Piollet, H.; Viallet, F.; Khalfallah, Y.; Chazot, G. Magnetic Resonance Imaging Evidence of Decreased Putamenal Iron Content in Idiopathic Parkinson’s Disease. Arch. Neurol. 1995, 52, 583–588. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Corcia, P.; Mucha, A.; Benzimra, S.; Mallet, C.; Gendrot, C.; Moreau, C.; Devos, D.; Piver, E.; Pagès, J.-C.; et al. Iron Metabolism Disturbance in a French Cohort of ALS Patients. BioMed Res. Int. 2014, 2014, 485723. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, M.; Brown Jr, R.H.; Rogers, J.T.; Cudkowicz, M.E. Serum Ferritin and Metal Levels as Risk Factors for Amyotrophic Lateral Sclerosis. Open Neurol. J. 2008, 2, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Núñez, M.T. Mitochondrial Iron Homeostasis and Its Dysfunctions in Neurodegenerative Disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Ferri, A.; Cozzolino, M.; Calabrese, L.; Rotilio, G. Neurodegeneration in Amyotrophic Lateral Sclerosis: The Role of Oxidative Stress and Altered Homeostasis of Metals. Brain Res. Bull. 2003, 61, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Rathore, K.I.; Schulz, K.; Ponka, P.; Arosio, P.; David, S. Dysregulation of Iron Homeostasis in the CNS Contributes to Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2009, 29, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primer 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Hochstrasser, H. Iron Metabolism in Parkinsonian Syndromes. Mov. Disord. 2006, 21, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria Mass Is Low in Mouse Substantia Nigra Dopamine Neurons: Implications for Parkinson’s Disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Sado, M.; Yamasaki, Y.; Iwanaga, T.; Onaka, Y.; Ibuki, T.; Nishihara, S.; Mizuguchi, H.; Momota, H.; Kishibuchi, R.; Hashimoto, T.; et al. Protective Effect against Parkinson’s Disease-Related Insults through the Activation of XBP1. Brain Res. 2009, 1257, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Cabuy, E.; Caroni, P. A Role for Motoneuron Subtype–Selective ER Stress in Disease Manifestations of FALS Mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative Stress and Neurodegeneration: The Involvement of Iron. BioMetals 2018, 31, 715–735. [Google Scholar] [CrossRef]
- Kim, Y.; Connor, J.R. The Roles of Iron and HFE Genotype in Neurological Diseases. Mol. Asp. Med. 2020, 75, 100867. [Google Scholar] [CrossRef]
- Batista-Nascimento, L.; Pimentel, C.; Andrade Menezes, R.; Rodrigues-Pousada, C. Iron and Neurodegeneration: From Cellular Homeostasis to Disease. Oxidative Med. Cell. Longev. 2012, 2012, 128647. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- De Souza, G.F.; Ribeiro, H.L.; De Sousa, J.C.; Heredia, F.F.; De Freitas, R.M.; Martins, M.R.A.; Gonçalves, R.P.; Pinheiro, R.F.; Magalhães, S.M.M. HFE Gene Mutation and Oxidative Damage Biomarkers in Patients with Myelodysplastic Syndromes and Its Relation to Transfusional Iron Overload: An Observational Cross-Sectional Study. BMJ Open 2015, 5, e006048. [Google Scholar] [CrossRef]
- Merryweather-Clarke, A.T.; Pointon, J.J.; Jouanolle, A.M.; Rochette, J.; Robson, K.J.H. Geography of HFEC282Y and H63D Mutations. Genet. Test. 2000, 4, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, K.K.; Cogswell, M.E.; Chang, J.C.; Caudill, S.P.; McQuillan, G.M.; Bowman, B.A.; Grummer-Strawn, L.M.; Sampson, E.J.; Khoury, M.J.; Gallagher, M.L. Prevalence of C282Y and H63D Mutations in the Hemochromatosis (HFE) Gene in the United States. JAMA 2001, 285, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Sampietro, M.; Caputo, L.; Casatta, A.; Meregalli, M.; Pellagatti, A.; Tagliabue, J.; Annoni, G.; Vergani, C. The Hemochromatosis Gene Affects the Age of Onset of Sporadic Alzheimer’s Disease. Neurobiol. Aging 2001, 22, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Berlin, D.; Chong, G.; Chertkow, H.; Bergman, H.; Phillips, N.A.; Schipper, H.M. Evaluation of HFE (Hemochromatosis) Mutations as Genetic Modifiers in Sporadic AD and MCI. Neurobiol. Aging 2004, 25, 465–474. [Google Scholar] [CrossRef]
- Halling, J.; Petersen, M.S.; Grandjean, P.; Weihe, P.; Brosen, K. Genetic Predisposition to Parkinson’s Disease: CYP2D6 and HFE in the Faroe Islands. Pharmacogenet. Genom. 2008, 18, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Borie, C.; Gasparini, F.; Verpillat, P.; Bonnet, A.-M.; Agid, Y.; Hetet, G.; Brice, A.; Dürr, A.; Grandchamp, B. Association Study between Iron-Related Genes Polymorphisms and Parkinson’s Disease. J. Neurol. 2002, 249, 801–804. [Google Scholar] [CrossRef]
- Greco, V.; De Marco, E.V.; Rocca, F.E.; Annesi, F.; Civitelli, D.; Provenzano, G.; Tarantino, P.; Scornaienchi, V.; Pucci, F.; Salsone, M.; et al. Association Study between Four Polymorphisms in the HFE, TF and TFR Genes and Parkinson’s Disease in Southern Italy. Neurol. Sci. 2011, 32, 525–527. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Bras, J.M.; Santana, I.; Januario, C.; Santiago, B.; Morgadinho, A.S.; Ribeiro, M.H.; Hardy, J.; Singleton, A.; Oliveira, C. Association of HFE Common Mutations with Parkinson’s Disease, Alzheimer’s Disease and Mild Cognitive Impairment in a Portuguese Cohort. BMC Neurol. 2006, 6, 24. [Google Scholar] [CrossRef]
- Akbas, N.; Hochstrasser, H.; Deplazes, J.; Tomiuk, J.; Bauer, P.; Walter, U.; Behnke, S.; Riess, O.; Berg, D. Screening for Mutations of the HFE Gene in Parkinson’s Disease Patients with Hyperechogenicity of the Substantia Nigra. Neurosci. Lett. 2006, 407, 16–19. [Google Scholar] [CrossRef]
- Biasiotto, G.; Goldwurm, S.; Finazzi, D.; Tunesi, S.; Zecchinelli, A.; Sironi, F.; Pezzoli, G.; Arosio, P. HFE Gene Mutations in a Population of Italian Parkinson’s Disease Patients. Parkinsonism Relat. Disord. 2008, 14, 426–430. [Google Scholar] [CrossRef]
- Dekker, M.C.J.; Giesbergen, P.C.; Njajou, O.T.; van Swieten, J.C.; Hofman, A.; Breteler, M.M.B.; van Duijn, C.M. Mutations in the Hemochromatosis Gene (HFE), Parkinson’s Disease and Parkinsonism. Neurosci. Lett. 2003, 348, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Xu, H.; Jiang, H.; Xie, J. The Association between the C282Y and H63D Polymorphisms of HFE Gene and the Risk of Parkinson’s Disease: A Meta-Analysis. Neurosci. Lett. 2015, 595, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.W.; Trojano, M.L.; Lewis, M.M.; Du, G.; Chen, H.; Brown, G.L.; Jellen, L.C.; Song, I.; Neely, E.; Kong, L.; et al. HFE H63D Limits Nigral Vulnerability to Paraquat in Agricultural Workers. Toxicol. Sci. 2021, 181, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Goodall, E.F.; Greenway, M.J.; van Marion, I.; Carroll, C.B.; Hardiman, O.; Morrison, K.E. Association of the H63D Polymorphism in the Hemochromatosis Gene with Sporadic ALS. Neurology 2005, 65, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Sutedja, N.A.; Sinke, R.J.; Vught, P.W.J.V.; der Linden, M.W.V.; Wokke, J.H.J.; Duijn, C.M.V.; Njajou, O.T.; der Schouw, Y.T.V.; Veldink, J.H.; Berg, L.H.V. den The Association between H63D Mutations in HFE and Amyotrophic Lateral Sclerosis in a Dutch Population. Arch. Neurol. 2007, 64, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Restagno, G.; Lombardo, F.; Ghiglione, P.; Calvo, A.; Cocco, E.; Sbaiz, L.; Mutani, R.; Chiò, A. HFE H63D Polymorphism Is Increased in Patients with Amyotrophic Lateral Sclerosis of Italian Origin. J. Neurol. Neurosurg. Psychiatry 2007, 78, 327. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Mora, G.; Sabatelli, M.; Caponnetto, C.; Lunetta, C.; Traynor, B.J.; Johnson, J.O.; Nalls, M.A.; Calvo, A.; Moglia, C.; et al. HFE p.H63D Polymorphism Does Not Influence ALS Phenotype and Survival. Neurobiol. Aging 2015, 36, 2906.e7–2906.e11. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-S.; Lee, S.; Simmons, Z.; Boyer, P.; Scott, K.; Liu, W.; Connor, J. Increased Incidence of the Hfe Mutation in Amyotrophic Lateral Sclerosis and Related Cellular Consequences. J. Neurol. Sci. 2004, 227, 27–33. [Google Scholar] [CrossRef]
- Yen, A.A.; Simpson, E.P.; Henkel, J.S.; Beers, D.R.; Appel, S.H. HFE Mutations Are Not Strongly Associated with Sporadic ALS. Neurology 2004, 62, 1611–1612. [Google Scholar] [CrossRef]
- van Rheenen, W.; Diekstra, F.P.; van Doormaal, P.T.C.; Seelen, M.; Kenna, K.; McLaughlin, R.; Shatunov, A.; Czell, D.; van Es, M.A.; van Vught, P.W.J.; et al. H63D Polymorphism in HFE Is Not Associated with Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2013, 34, 1517.e5–1517.e7. [Google Scholar] [CrossRef]
- Praline, J.; Blasco, H.; Vourc’h, P.; Rat, V.; Gendrot, C.; Camu, W.; Andres, C.R. Study of the HFE Gene Common Polymorphisms in French Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2012, 317, 58–61. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Wang, W.; Qi, X.L.; Tang, Z.Y. Mutations in the HFE Gene and Sporadic Amyotrophic Lateral Sclerosis Risk: A Meta-Analysis of Observational Studies. Braz. J. Med. Biol. Res. 2014, 47, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Lee, S.Y.; Mitchell, R.M.; Stephens, H.E.; Simmons, Z.; Connor, J.R. H63D HFE Polymorphisms Are Associated with Increased Disease Duration and Decreased Muscle Superoxide Dismutase-1 Expression in Amyotrophic Lateral Sclerosis Patients. Muscle Nerve 2013, 48, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Nandar, W.; Neely, E.B.; Unger, E.; Connor, J.R. A Mutation in the HFE Gene Is Associated with Altered Brain Iron Profiles and Increased Oxidative Stress in Mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Song, I.Y.; Snyder, A.M.; Kim, Y.; Neely, E.B.; Wade, Q.W.; Connor, J.R. The Nrf2-Mediated Defense Mechanism Associated with HFE Genotype Limits Vulnerability to Oxidative Stress-Induced Toxicity. Toxicology 2020, 441, 152525. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Stahl, M.C.; Huang, X.; Connor, J.R. H63D Variant of the Homeostatic Iron Regulator (HFE) Gene Alters α-Synuclein Expression, Aggregation, and Toxicity. J. Neurochem. 2020, 155, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.M.; Meadowcroft, M.D.; Neely, E.B.; Snyder, A.M.; Purnell, C.J.; Wright, J.; Lamendella, R.; Nandar, W.; Huang, X.; Connor, J.R. HFE Genotype Restricts the Response to Paraquat in a Mouse Model of Neurotoxicity. J. Neurochem. 2018, 145, 299–311. [Google Scholar] [CrossRef]
- Mattson, M.P. Hormesis Defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Mao, L.; Franke, J. Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection. Int. J. Mol. Sci. 2013, 14, 13109–13128. [Google Scholar] [CrossRef]
- Zimmermann, A.; Bauer, M.A.; Kroemer, G.; Madeo, F.; Carmona-Gutierrez, D. When Less Is More: Hormesis against Stress and Disease. Microb. Cell 2010, 1, 150–153. [Google Scholar] [CrossRef]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, Oxidative Stress and Hormesis. Ageing Res. Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A Novel MHC Class I-like Gene Is Mutated in Patients with Hereditary Haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Cardoso, C.S.; Sousa, M.D. HFE, the MHC and Hemochromatosis: Paradigm for an Extended Function for MHC Class I. Tissue Antigens 2003, 61, 263–275. [Google Scholar] [CrossRef]
- Waheed, A.; Parkkila, S.; Saarnio, J.; Fleming, R.E.; Zhou, X.Y.; Tomatsu, S.; Britton, R.S.; Bacon, B.R.; Sly, W.S. Association of HFE Protein with Transferrin Receptor in Crypt Enterocytes of Human Duodenum. Proc. Natl. Acad. Sci. USA 1999, 96, 1579–1584. [Google Scholar] [CrossRef]
- Pietrangelo, A. Physiology of Iron Transport and the Hemochromatosis Gene. Am. J. Physiol.-Gastrointest. Liver Physiol. 2002, 282, G403–G414. [Google Scholar] [CrossRef]
- Lebrón, J.A.; Bennett, M.J.; Vaughn, D.E.; Chirino, A.J.; Snow, P.M.; Mintier, G.A.; Feder, J.N.; Bjorkman, P.J. Crystal Structure of the Hemochromatosis Protein HFE and Characterization of Its Interaction with Transferrin Receptor. Cell 1998, 93, 111–123. [Google Scholar] [CrossRef]
- Feder, J.N.; Penny, D.M.; Irrinki, A.; Lee, V.K.; Lebrón, J.A.; Watson, N.; Tsuchihashi, Z.; Sigal, E.; Bjorkman, P.J.; Schatzman, R.C. The Hemochromatosis Gene Product Complexes with the Transferrin Receptor and Lowers Its Affinity for Ligand Binding. Proc. Natl. Acad. Sci. USA 1998, 95, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Giannetti, A.M.; Björkman, P.J. HFE and Transferrin Directly Compete for Transferrin Receptor in Solution and at the Cell Surface. J. Biol. Chem. 2004, 279, 25866–25875. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.N.; Irrinki, A.; Feder, J.N.; Enns, C.A. Co-Trafficking of HFE, a Nonclassical Major Histocompatibility Complex Class I Protein, with the Transferrin Receptor Implies a Role in Intracellular Iron Regulation. J. Biol. Chem. 1998, 273, 22068–22074. [Google Scholar] [CrossRef] [PubMed]
- Lebrón, J.A.; West, A.P.; Bjorkman, P.J. The Hemochromatosis Protein HFE Competes with Transferrin for Binding to the Transferrin Receptor11. J. Mol. Biol. 1999, 294, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G. Genetic Mechanisms and Modifying Factors in Hereditary Hemochromatosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.; Imperatore, G.; McDonnell, S.M.; Baron, R.C.; Khoury, M.J. Contribution of Different HFE Genotypes to Iron Overload Disease: A Pooled Analysis. Genet. Med. 2000, 2, 271–277. [Google Scholar] [CrossRef]
- Barry, E.; Derhammer, T.; Elsea, S.H. Prevalence of Three Hereditary Hemochromatosis Mutant Alleles in the Michigan Caucasian Population. Public Health Genom. 2005, 8, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.A.; Carter, K.; Darke, C.; Guttridge, M.G.; Ravine, D.; Hutton, R.D.; Napier, J.A.; Worwood, M. HFE Mutations, Iron Deficiency and Overload in 10 500 Blood Donors. Br. J. Haematol. 2001, 114, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Acton, R.T.; Barton, J.C.; Snively, B.M.; McLaren, C.E.; Adams, P.C.; Harris, E.L.; Speechley, M.R.; McLaren, G.D.; Dawkins, F.W.; Leiendecker-Foster, C.; et al. Geographic and Racial/Ethnic Differences in HFE Mutation Frequencies in the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Ethn. Dis. 2006, 16, 815–821. [Google Scholar] [CrossRef]
- Hermine, O.; Dine, G.; Genty, V.; Marquet, L.-A.; Fumagalli, G.; Tafflet, M.; Guillem, F.; Van Lierde, F.; Rousseaux-Blanchi, M.-P.; Palierne, C.; et al. Eighty Percent of French Sport Winners in Olympic, World and Europeans Competitions Have Mutations in the Hemochromatosis HFE Gene. Biochimie 2015, 119, 1–5. [Google Scholar] [CrossRef]
- Semenova, E.A.; Miyamoto-Mikami, E.; Akimov, E.B.; Al-Khelaifi, F.; Murakami, H.; Zempo, H.; Kostryukova, E.S.; Kulemin, N.A.; Larin, A.K.; Borisov, O.V.; et al. The Association of HFE Gene H63D Polymorphism with Endurance Athlete Status and Aerobic Capacity: Novel Findings and a Meta-Analysis. Eur. J. Appl. Physiol. 2020, 120, 665–673. [Google Scholar] [CrossRef]
- Thakkar, D.; Sicova, M.; Guest, N.S.; Garcia-Bailo, B.; El-Sohemy, A. HFE Genotype and Endurance Performance in Competitive Male Athletes. Med. Sci. Sports Exerc. 2021, 53, 1385–1390. [Google Scholar] [CrossRef]
- Chicharro, J.; Hoyos, J.; Gomez-Gallego, F.; Villa, J.; Bandres, F.; Celaya, P.; Jimenez, F.; Alonso, J.; Cordova, A.; Lucia, A. Mutations in the Hereditary Haemochromatosis Gene HFE in Professional Endurance Athletes. Br. J. Sports Med. 2004, 38, 418–421. [Google Scholar] [CrossRef]
- Deugnier, Y.; Loréal, O.; Carré, F.; Duvallet, A.; Zoulim, F.; Vinel, J.P.; Paris, J.C.; Blaison, D.; Moirand, R.; Turlin, B.; et al. Increased Body Iron Stores in Elite Road Cyclists. Med. Sci. Sports Exerc. 2002, 34, 876–880. [Google Scholar] [CrossRef]
- Yokoyama, J.S.; Bonham, L.W.; Sears, R.L.; Klein, E.; Karydas, A.; Kramer, J.H.; Miller, B.L.; Coppola, G. Decision Tree Analysis of Genetic Risk for Clinically Heterogeneous Alzheimer’s Disease. BMC Neurol. 2015, 15, 47. [Google Scholar] [CrossRef]
- Pulliam, J.F.; Jennings, C.D.; Kryscio, R.J.; Davis, D.G.; Wilson, D.; Montine, T.J.; Schmitt, F.A.; Markesbery, W.R. Association of HFE Mutations with Neurodegeneration and Oxidative Stress in Alzheimer’s Disease and Correlation with APOE. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 119B, 48–53. [Google Scholar] [CrossRef]
- Percy, M.; Moalem, S.; Garcia, A.; Somerville, M.J.; Hicks, M.; Andrews, D.; Azad, A.; Schwarz, P.; Zavareh, R.B.; Birkan, R.; et al. Involvement of ApoE E4 and H63D in Sporadic Alzheimer’s Disease in a Folate-Supplemented Ontario Population. J. Alzheimer’s Dis. 2008, 14, 69–84. [Google Scholar] [CrossRef]
- Blázquez, L.; De Juan, D.; Ruiz-Martínez, J.; Emparanza, J.I.; Sáenz, A.; Otaegui, D.; Sistiaga, A.; Martínez-Lage, P.; Lamet, I.; Samaranch, L.; et al. Genes Related to Iron Metabolism and Susceptibility to Alzheimer’s Disease in Basque Population. Neurobiol. Aging 2007, 28, 1941–1943. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhao, L.; Fan, J.; Lian, X.-G.; Ye, J.; Wu, L.; Lin, H. Association between HFE Polymorphisms and Susceptibility to Alzheimer’s Disease: A Meta-Analysis of 22 Studies Including 4,365 Cases and 8,652 Controls. Mol. Biol. Rep. 2012, 39, 3089–3095. [Google Scholar] [CrossRef]
- Correia, A.P.; Pinto, J.P.; Dias, V.; Mascarenhas, C.; Almeida, S.; Porto, G. CAT53 and HFE Alleles in Alzheimer’s Disease: A Putative Protective Role of the C282Y HFE Mutation. Neurosci. Lett. 2009, 457, 129–132. [Google Scholar] [CrossRef]
- Lehmann, D.J.; Schuur, M.; Warden, D.R.; Hammond, N.; Belbin, O.; Kölsch, H.; Lehmann, M.G.; Wilcock, G.K.; Brown, K.; Kehoe, P.G.; et al. Transferrin and HFE Genes Interact in Alzheimer’s Disease Risk: The Epistasis Project. Neurobiol. Aging 2012, 33, 202.e1–202.e13. [Google Scholar] [CrossRef] [PubMed]
- Combarros, O.; García-Román, M.; Fontalba, A.; Fernández-Luna, J.L.; Llorca, J.; Infante, J.; Berciano, J. Interaction of the H63D Mutation in the Hemochromatosis Gene with the Apolipoprotein E Epsilon 4 Allele Modulates Age at Onset of Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2003, 15, 151–154. [Google Scholar] [CrossRef]
- Alizadeh, B.Z.; Njajou, O.T.; Millán, M.R.; Hofman, A.; Breteler, M.M.; van Duijn, C.M. HFE Variants, APOE and Alzheimer’s Disease: Findings from the Population-Based Rotterdam Study. Neurobiol. Aging 2009, 30, 330–332. [Google Scholar] [CrossRef]
- Kauwe, J.; Bertelsen, S.; Mayo, K.; Cruchaga, C.; Abraham, R.; Hollingworth, P.; Harold, D.; Owen, M.; Williams, J.; Lovestone, S.; et al. Suggestive Synergy between Genetic Variants in TF and HFE as Risk Factors for Alzheimer’s Disease. Am. J. Med. Genet. 2010, 153B, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Robson, K.; Lehmann, D.; Wimhurst, V.; Livesey, K.; Combrinck, M.; Merryweather-Clar, A.T.; Warden, D.; Smith, A. Synergy between the C2 Allele of Transferrin and the C282Y Allele of the Haemochromatosis Gene (HFE) as Risk Factors for Developing Alzheimer’s Disease. J. Med. Genet. 2004, 41, 261–265. [Google Scholar] [CrossRef]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild Cognitive Impairment: Clinical Characterization and Outcome. Arch. Neurol. 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Avila-gomez, I.C.; Jiménez-del-rio, M.; Lopera-restrepo, F.; Velez-pardo, C. Association between HFE 187 C>G (H63D) Mutation and Early-Onset Familial Alzheimer’s Disease PSEN-1 839A>C (E280A) Mutation. Ann. Hematol. 2008, 87, 671–673. [Google Scholar] [CrossRef]
- Candore, G.; Licastro, F.; Chiappelli, M.; Franceschi, C.; Lio, D.; Rita Balistreri, C.; Piazza, G.; Colonna-Romano, G.; Grimaldi, L.M.; Caruso, C. Association between the HFE Mutations and Unsuccessful Ageing: A Study in Alzheimer’s Disease Patients from Northern Italy. Mech. Ageing Dev. 2003, 124, 525–528. [Google Scholar] [CrossRef]
- Corder, E.H.; Beaumont, H. Susceptibility Groups for Alzheimer’s Disease (OPTIMA Cohort): Integration of Gene Variants and Biochemical Factors. Mech. Ageing Dev. 2007, 128, 76–82. [Google Scholar] [CrossRef]
- Giambattistelli, F.; Bucossi, S.; Salustri, C.; Panetta, V.; Mariani, S.; Siotto, M.; Ventriglia, M.; Vernieri, F.; Dell’Acqua, M.L.; Cassetta, E.; et al. Effects of Hemochromatosis and Transferrin Gene Mutations on Iron Dyshomeostasis, Liver Dysfunction and on the Risk of Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1633–1641. [Google Scholar] [CrossRef]
- Lleo, A.; Blesa, R.; Angelopoulos, C.; Pastor-Rubio, P.; Villa, M.; Oliva, R.; Bufill, E. Transferrin C2 Allele, Haemochromatosis Gene Mutations, and Risk for Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2002, 72, 820–821. [Google Scholar] [CrossRef]
- Mariani, S.; Ventriglia, M.; Simonelli, I.; Spalletta, G.; Bucossi, S.; Siotto, M.; Assogna, F.; Melgari, J.M.; Vernieri, F.; Squitti, R. Effects of Hemochromatosis and Transferrin Gene Mutations on Peripheral Iron Dyshomeostasis in Mild Cognitive Impairment and Alzheimer’s and Parkinson’s Diseases. Front. Aging Neurosci. 2013, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Vance, E.; Gonzalez Murcia, J.D.; Miller, J.B.; Staley, L.; Crane, P.K.; Mukherjee, S.; Kauwe, J.S.K. Failure to Detect Synergy between Variants in Transferrin and Hemochromatosis and Alzheimer’s Disease in Large Cohort. Neurobiol. Aging 2020, 89, 142.e9–142.e12. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.J.; Walsh, S.L.; Harrington, H.J.; Walsh, C.H. Concurrent Hereditary Haemochromatosis and Idiopathic Parkinson’s Disease: A Case Report Series. J. Neurol. Neurosurg. Psychiatry 2004, 75, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Aamodt, A.H.; Stovner, L.J.; Thorstensen, K.; Lydersen, S.; White, L.R.; Aasly, J.O. Prevalence of Haemochromatosis Gene Mutations in Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 315–317. [Google Scholar] [CrossRef]
- Buchanan, D.D.; Silburn, P.A.; Chalk, J.B.; Le Couteur, D.G.; Mellick, G.D. The Cys282Tyr Polymorphism in the HFE Gene in Australian Parkinson’s Disease Patients. Neurosci. Lett. 2002, 327, 91–94. [Google Scholar] [CrossRef]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic Criteria for Parkinson Disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Distante, S.; Berg, J.P.; Lande, K.; Haug, E.; Bell, H. High Prevalence of the Hemochromatosis-Associated Cys282Tyr HFE Gene Mutation in a Healthy Norwegian Population in the City of Oslo, and Its Phenotypic Expression. Scand. J. Gastroenterol. 1999, 34, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Li, J.; Jian, S.; Li, B.; Huang, Z.; Shu, L.; Zhang, Y. Quantitative and Causal Analysis for Inflammatory Genes and the Risk of Parkinson’s Disease. Front. Immunol. 2023, 14, 1119315. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Bandres-Ciga, S.; Alcantud, J.L.; Ruz, C.; Postuma, R.B.; Gan-Or, Z. Common and Rare Variants in HFE Are Not Associated with Parkinson’s Disease in Europeans. Neurobiol. Aging 2021, 107, 174–177. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lu, X.; Hu, J.; Xi, J.; Zhou, D.; Shang, H.; Liu, L.; Zhou, H.; Yan, B.; Yu, L.; et al. H63D Polymorphism in the Hemochromatosis Gene Is Associated with Sporadic Amyotrophic Lateral Sclerosis in China. Eur. J. Neurol. 2011, 18, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Canosa, A.; Calvo, A.; Mora, G.; Moglia, C.; Brunetti, M.; Barberis, M.; Borghero, G.; Caponnetto, C.; Trojsi, F.; Spataro, R.; et al. The HFE p.H63D (p.His63Asp) Polymorphism Is a Modifier of ALS Outcome in Italian and French Patients with SOD1 Mutations. Biomedicines 2023, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- Kalpouzos, G.; Mangialasche, F.; Falahati, F.; Laukka, E.J.; Papenberg, G. Contributions of HFE Polymorphisms to Brain and Blood Iron Load, and Their Links to Cognitive and Motor Function in Healthy Adults. Neuropsychopharmacol. Rep. 2021, 41, 393–404. [Google Scholar] [CrossRef]
- Ellervik, C.; Birgens, H.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Hemochromatosis Genotypes and Risk of 31 Disease Endpoints: Meta-Analyses Including 66,000 Cases and 226,000 Controls. Hepatology 2007, 46, 1071–1080. [Google Scholar] [CrossRef]
- Atkins, J.L.; Pilling, L.C.; Heales, C.J.; Savage, S.; Kuo, C.-L.; Kuchel, G.A.; Steffens, D.C.; Melzer, D. Hemochromatosis Mutations, Brain Iron Imaging, and Dementia in the UK Biobank Cohort. J. Alzheimer’s Dis. 2021, 79, 1203–1211. [Google Scholar] [CrossRef]
- Gebril, O.H.; Kirby, J.; Savva, G.; Brayne, C.; Ince, P.G. HFE H63D, C282Y and AGTR1 A1166C Polymorphisms and Brain White Matter Lesions in the Aging Brain. J. Neurogenet. 2011, 25, 7–14. [Google Scholar] [CrossRef]
- Percy, M.; Somerville, M.J.; Hicks, M.; Colelli, T.; Wright, E.; Kitaygorodsky, J.; Jiang, A.; Ho, V.; Parpia, A.; Wong, M.K.; et al. Risk Factors for Development of Dementia in a Unique Six-Year Cohort Study. I. An Exploratory, Pilot Study of Involvement of the E4 Allele of Apolipoprotein E, Mutations of the Hemochromatosis-HFE Gene, Type 2 Diabetes, and Stroke. J. Alzheimer’s Dis. 2013, 38, 907–922. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How Increased Oxidative Stress Promotes Longevity and Metabolic Health: The Concept of Mitochondrial Hormesis (Mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, A. Neurohormetic Phytochemicals: Low-Dose Toxins That Induce Adaptive Neuronal Stress Responses. Trends Neurosci. 2006, 29, 632–639. [Google Scholar] [CrossRef]
- Murugaiyah, V.; Mattson, M.P. Neurohormetic Phytochemicals: An Evolutionary—Bioenergetic Perspective. Neurochem. Int. 2015, 89, 271–280. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Tomatsu, S.; Fleming, R.E.; Parkkila, S.; Waheed, A.; Jiang, J.; Fei, Y.; Brunt, E.M.; Ruddy, D.A.; Prass, C.E.; et al. HFE Gene Knockout Produces Mouse Model of Hereditary Hemochromatosis. Proc. Natl. Acad. Sci. USA 1998, 95, 2492–2497. [Google Scholar] [CrossRef]
- Ye, Q.; Trivedi, M.; Zhang, Y.; Böhlke, M.; Alsulimani, H.; Chang, J.; Maher, T.; Deth, R.; Kim, J. Brain Iron Loading Impairs DNA Methylation and Alters GABAergic Function in Mice. FASEB J. 2019, 33, 2460–2471. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Kim, J. Mutation in HFE Gene Decreases Manganese Accumulation and Oxidative Stress in the Brain after Olfactory Manganese Exposure. Met. Integr. Biomet. Sci. 2016, 8, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Nandar, W.; Neely, E.B.; Simmons, Z.; Connor, J.R. H63D HFE Genotype Accelerates Disease Progression in Animal Models of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Lawless, M.W.; Mankan, A.K.; White, M.; O’Dwyer, M.J.; Norris, S. Expression of Hereditary Hemochromatosis C282Y HFE Protein in HEK293 Cells Activates Specific Endoplasmic Reticulum Stress Responses. BMC Cell Biol. 2007, 8, 30. [Google Scholar] [CrossRef]
- Lee, S.Y.; Patton, S.M.; Henderson, R.J.; Connor, J.R. Consequences of Expressing Mutants of the Hemochromatosis Gene (HFE) into a Human Neuronal Cell Line Lacking Endogenous HFE. FASEB J. 2007, 21, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Mairuae, N.; Ii, E.C.H.; Cheepsunthorn, P.; Lee, S.Y.; Connor, J.R. The H63D HFE Gene Variant Promotes Activation of the Intrinsic Apoptotic Pathway via Mitochondria Dysfunction Following β-Amyloid Peptide Exposure. J. Neurosci. Res. 2010, 88, 3079–3089. [Google Scholar] [CrossRef] [PubMed]
- Ali-Rahmani, F.; Hengst, J.A.; Connor, J.R.; Schengrund, C.-L. Effect of HFE Variants on Sphingolipid Expression by SH-SY5Y Human Neuroblastoma Cells. Neurochem. Res. 2011, 36, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.C.; Lee, S.Y.; Mairuae, N.; Simmons, Z.; Connor, J.R. Expression of the HFE Allelic Variant H63D in SH-SY5Y Cells Affects Tau Phosphorylation at Serine Residues. Neurobiol. Aging 2011, 32, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.M.; Lee, S.Y.; Simmons, Z.; Connor, J.R. HFE Polymorphisms Affect Cellular Glutamate Regulation. Neurobiol. Aging 2011, 32, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants Prevent Health-Promoting Effects of Physical Exercise in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear Factor E2-Related Factor 2-Dependent Antioxidant Response Element Activation by Tert-Butylhydroquinone and Sulforaphane Occurring Preferentially in Astrocytes Conditions Neurons against Oxidative Insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef]
- Xu, J.; Huang, G.; Zhang, K.; Sun, J.; Xu, T.; Li, R.; Tao, H.; Xu, W. Nrf2 Activation in Astrocytes Contributes to Spinal Cord Ischemic Tolerance Induced by Hyperbaric Oxygen Preconditioning. J. Neurotrauma 2014, 31, 1343–1353. [Google Scholar] [CrossRef]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Administration of the Nrf2–ARE Activators Sulforaphane and Carnosic Acid Attenuates 4-Hydroxy-2-Nonenal-Induced Mitochondrial Dysfunction Ex Vivo. Free Radic. Biol. Med. 2013, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl Fumarate Mediates Nrf2-Dependent Mitochondrial Biogenesis in Mice and Humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef]
- O’Mealey, G.B.; Plafker, K.S.; Berry, W.L.; Janknecht, R.; Chan, J.Y.; Plafker, S.M. A PGAM5–KEAP1–Nrf2 Complex Is Required for Stress-Induced Mitochondrial Retrograde Trafficking. J. Cell Sci. 2017, 130, 3467–3480. [Google Scholar] [CrossRef]
- Bento-Pereira, C.; Dinkova-Kostova, A.T. Activation of Transcription Factor Nrf2 to Counteract Mitochondrial Dysfunction in Parkinson’s Disease. Med. Res. Rev. 2021, 41, 785–802. [Google Scholar] [CrossRef]
- Cabantchik, Z.I. Labile Iron in Cells and Body Fluids: Physiology, Pathology, and Pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall Moscon, S.L.; Connor, J.R. HFE Mutations in Neurodegenerative Disease as a Model of Hormesis. Int. J. Mol. Sci. 2024, 25, 3334. https://doi.org/10.3390/ijms25063334
Marshall Moscon SL, Connor JR. HFE Mutations in Neurodegenerative Disease as a Model of Hormesis. International Journal of Molecular Sciences. 2024; 25(6):3334. https://doi.org/10.3390/ijms25063334
Chicago/Turabian StyleMarshall Moscon, Savannah L., and James R. Connor. 2024. "HFE Mutations in Neurodegenerative Disease as a Model of Hormesis" International Journal of Molecular Sciences 25, no. 6: 3334. https://doi.org/10.3390/ijms25063334