
Beyond Quiescent and Active: Intermediate Microglial Transcriptomic States in a Mouse Model of Down Syndrome


Abstract
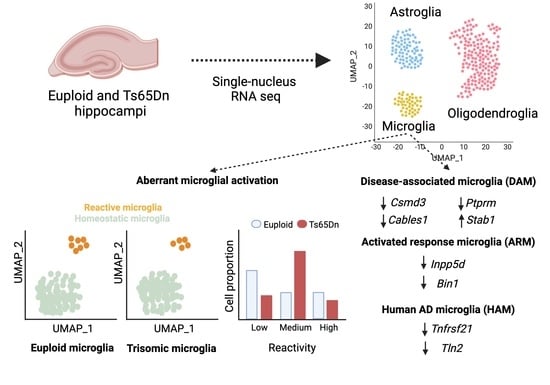
:
1. Introduction
2. Results
2.1. Unbiased Identification of Different Glial Subtypes in WT and Ts65Dn Hippocampus
2.2. Trisomic Glial Cells Exhibit Cell-Type-Specific Transcriptomic Alterations
2.3. More than 60% of Trisomic Microglial Cells Are in an Intermediate Reactive State
3. Discussion
Limitations
4. Materials and Methods
4.1. Animals
4.2. Histology
4.2.1. Immunohistochemistry
4.2.2. Cell Counting
4.3. Single Nucleus RNA Sequencing
4.3.1. Nucleus Isolation
4.3.2. 10x Single-Cell Barcoding, Library Preparation, and Sequencing
4.3.3. 10x Data Pre-Processing
4.3.4. Batch Correction and Scaling Data Matrix
4.4. Dimensionality Reduction, Clustering and Visualization
4.5. Identification of Marker Genes within Every Cluster
4.6. Identification of Differentially Expressed Genes between WT and Ts65Dn
4.7. Gene Set Enrichment
4.8. Cellular Proportion
4.9. Disease-Associated Microglia (DAM) Markers
4.10. Pseudotime Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ross, M.H.; Galaburda, A.M.; Kemper, T.L. Down’s syndrome: Is there a decreased population of neurons? Neurology 1984, 34, 909–916. [Google Scholar] [CrossRef]
- Guidi, S.; Bonasoni, P.; Ceccarelli, C.; Santini, D.; Gualtieri, F.; Ciani, E.; Bartesaghi, R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008, 18, 180–197. [Google Scholar] [CrossRef]
- Pinto, B.; Morelli, G.; Rastogi, M.; Savardi, A.; Fumagalli, A.; Petretto, A.; Petretto, A.; Bartolucci, M.; Varea, E.; Catelani, T.; et al. Rescuing Over-activated Microglia Restores Cognitive Performance in Juvenile Animals of the Dp(16) Mouse Model of Down Syndrome. Neuron 2020, 108, 887–904.e12. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, B.; Audinat, E.; Bechmann, I.; Bennet, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflam. 2021, 18, 258. [Google Scholar] [CrossRef]
- Provenzano, F.; Pérez, M.J.; Deleidi, M. Redefining Microglial Identity in Health and Disease at Single-Cell Resolution. Trends Mol. Med. 2021, 27, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.; Modrusan, Z.; Beach, T.; Serrano, G.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.; Song, A.; Liu, P.; Lin, J.-P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Wierzba-Bobrowicz, T.; Lewandowska, E.; Schmidt-Sidor, B.; Gwiazda, E. The comparison of microglia maturation in CNS of normal human fetuses and fetuses with Down’s syndrome. Folia Neuropathol. 1999, 37, 227–234. [Google Scholar]
- Xue, Q.S.; Streit, W.J. Microglial pathology in Down syndrome. Acta Neuropathol. 2011, 122, 455–466. [Google Scholar] [CrossRef]
- Flores-Aguilar, L.; Iulita, M.F.; Kovecses, O.; Torres, M.D.; Levi, S.M.; Zhang, Y.; Askenazi, M.; Wisniewski, T.; Busciglio, J.; Cuello, A.C. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 2020, 143, 3653–3671. [Google Scholar] [CrossRef]
- Martini, A.C.; Helman, A.M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Schmitt, F.A.; Head, E. Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down syndrome. Alzheimer’s Dement Diagnosis. Assess Dis. Monit. 2020, 12, e12113. [Google Scholar] [CrossRef]
- Colton, C.A.; Yao, J.; Taffs, R.E.; Keri, J.E.; Oster-Granite, M.L. Abnormal production of interleukin-1 by microglia from trisomy 16 mice. Neurosci. Lett. 1991, 132, 270–274. [Google Scholar] [CrossRef]
- Palmer, C.R.; Liu, C.S.; Romanow, W.J.; Lee, M.H.; Chun, J. Altered cell and RNA isoform diversity in aging down syndrome brains. Proc. Natl. Acad. Sci. USA 2021, 118, e2114326118. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Hurban, J.; Helman, A.M.; Sudduth, T.L.; McCarty, K.L.; Beckett, T.L.; Ferrell, J.C.; Murphy, M.P.; Abner, E.L.; Schmitt, F.A.; et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; Vidal, V.; García-Cerro, S.; Narcís, J.O.; Llorens-Martín, M.; Corrales, A.; Lantigua, S.; Iglesias, M.; Merino, J.; Merino, R.; et al. Anti-IL17 treatment ameliorates Down syndrome phenotypes in mice. Brain Behav. Immun. 2018, 73, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Illouz, T.; Madar, R.; Biragyn, A.; Okun, E. Restoring microglial and astroglial homeostasis using DNA immunization in a Down Syndrome mouse model. Brain Behav. Immun. 2019, 75, 163–180. [Google Scholar] [CrossRef]
- Lockrow, J.; Boger, H.; Bimonte-Nelson, H.; Granholm, A.C. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav. Brain Res. 2011, 221, 610–622. [Google Scholar] [CrossRef]
- Hamlett, E.D.; Hjorth, E.; Ledreux, A.; Gilmore, A.; Schultzberg, M.; Granholm, A.C. RvE1 treatment prevents memory loss and neuroinflammation in the Ts65Dn mouse model of Down syndrome. Glia 2020, 68, 1347–1360. [Google Scholar] [CrossRef]
- Gupta, M.; Dhanasekaran, A.R.; Gardiner, K.J. Mouse models of Down syndrome: Gene content and consequences. Mamm. Genome 2016, 27, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; Flórez, J.; Martínez-Cué, C. Mouse models of down syndrome as a tool to unravel the causes of mental disabilities. Neural Plast. 2012, 2012, 584071. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Kang, H.J.; Tyler, W.A.; Silbereis, J.C.; Cheng, F.; Zhu, Y.; Pletikos, M.; Jankovic-Rapan, L.; Cramer, N.P.; Galdzicki, Z.; et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron 2016, 89, 1208–1222. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhi, C.; Chen, D.; Cai, Z.; Jiang, X. Single-nucleus RNA sequencing reveals cell type-specific transcriptome alterations of Down syndrome hippocampus using the Dp16 mouse model. Genes. Genomics. 2023, 45, 1305–1315. [Google Scholar] [CrossRef]
- Qiu, J.J.; Liu, Y.N.; Wei, H.; Zeng, F.; Yan, J.B. Single-cell RNA sequencing of neural stem cells derived from human trisomic iPSCs reveals the abnormalities during neural differentiation of Down syndrome. Front. Mol. Neurosci. 2023, 16, 1137123. [Google Scholar] [CrossRef]
- Ponroy Bally, B.; Farmer, W.T.; Jones, E.V.; Jessa, S.; Kacerovsky, J.B.; Mayran, A.; Peng, H.; Lefebvre, J.L.; Drouin, J.; Hayer, A.; et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum. Mol. Genet. 2020, 29, 785–802. [Google Scholar] [CrossRef]
- Sierra, C.; Sabariego-Navarro, M.; Fernández-Blanco, Á.; Cruciani, S.; Zamora-Moratalla, A.; Novoa, E.M.; Dierssen, M. The lncRNA Snhg11, a new candidate contributing to neurogenesis, plasticity and memory deficits in Down syndrome. Mol. Psychiatry 2024, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Andrews, R.; Holmans, P.; Hill, M.; Taylor, P.R. Modest changes in Spi1 dosage reveal the potential for altered microglial function as seen in Alzheimer’s disease. Sci. Rep. 2021, 11, 14935. [Google Scholar] [CrossRef] [PubMed]
- Reifschneider, A.; Robinson, S.; Van Lengerich, B.; Gnörich, J.; Logan, T.; Heindl, S.; Vogt, M.A.; Weidinger, E.; Riedl, L.; Wind, K.; et al. Loss of TREM2 rescues hyperactivation of microglia, but not lysosomal deficits and neurotoxicity in models of progranulin deficiency. EMBO J. 2022, 41, e109108. [Google Scholar] [CrossRef] [PubMed]
- Sobue, A.; Komine, O.; Hara, Y.; Endo, F.; Mizoguchi, H.; Watanabe, S.; Murayama, S.; Saito, T.; Saido, T.C.; Sahara, N.; et al. Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gally, F.; Rao, D.M.; Schmitz, C.; Colvin, K.L.; Yeager, M.E.; Perraud, A.L. The TRPM2 ion channel contributes to cytokine hyperproduction in a mouse model of Down Syndrome. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 126–132. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Griffin, W.S.T. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflam. 2013, 10, 864. [Google Scholar] [CrossRef] [PubMed]
- Seol, S.; Kwon, J.; Kang, H.J. Cell type characterization of spatiotemporal gene co-expression modules in Down syndrome brain. iScience 2022, 26, 105884. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Shemer, A.; Levy-Efrati, L.; Gross, M.; Kim, J.S.; Engel, A.; David, E.; Chappell-Maor, L.; Grozovski, J.; Rotkopf, R.; et al. Microglial MHC class II is dispensable for experimental autoimmune encephalomyelitis and cuprizone-induced demyelination. Eur. J. Immunol. 2018, 48, 1308–1318. [Google Scholar] [CrossRef]
- Vázquez-Oliver, A.; Pérez-García, S.; Pizarro, N.; Molina-Porcel, L.; Torre R de la Maldonado, R.; Ozaita, A. Long-term decreased cannabinoid type-1 receptor activity restores specific neurological phenotypes in the Ts65Dn mouse model of Down syndrome. bioRxiv 2021. bioRxiv:2021.11.22.469296. [Google Scholar] [CrossRef]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White matter aging drives microglial diversity. Neuron 2021, 109, 1100–1117.e10. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Erny, D.; De Angelis, A.L.H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation function of microglia in the, C.N.S. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Thion, M.S.; Low, D.; Silvin, A.; Chen, J.; Grisel, P.; Schulte-Schrepping, J.; Blecher, R.; Ulas, T.; Squarzoni, P.; Hoeffel, G.; et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 2018, 172, 500–516.e16. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef]
- Song, W.; Li, Q.; Wang, T.; Li, Y.; Fan, T.; Zhang, J.; Wang, Q.; Pan, J.; Dong, Q.; Sun, Z.S.; et al. Putative complement control protein CSMD3 dysfunction impairs synaptogenesis and induces neurodevelopmental disorders. Brain Behav. Immun. 2022, 102, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Kálmán, J.; Juhász, A.; Laird, G.; Dickens, P.; Járdánházy, T.; Rimanóczy, Á.; Boncz, I.; Parry-Jones, W.L.; Janka, Z. Serum interleukin-6 levels correlate with the severity of dementia in Down syndrome and in Alzheimer’s disease. Acta Neurol. Scand. 1997, 96, 236–240. [Google Scholar] [CrossRef]
- Zhang, Y.; Che, M.; Yuan, J.; Yu, Y.; Cao, C.; Qin, X.Y.; Cheng, Y. Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: A meta-analysis. Oncotarget 2017, 8, 84489. [Google Scholar] [CrossRef]
- Wilcock, D.M. Neuroinflammation in the aging down syndrome brain; Lessons from Alzheimer’s disease. Curr. Gerontol. Geriatr. Res. 2012, 2012, 170276. [Google Scholar] [CrossRef]
- García, O.; Flores-Aguilar, L. Astroglial and microglial pathology in Down syndrome: Focus on Alzheimer’s disease. Front. Cell Neurosci. 2022, 16, 987212. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Martelotto, L.G. ‘Frankenstein’ Protocol for Nuclei Isolation from Fresh and Frozen Tissue for snRNAseq. Protocols. io 2020. [Google Scholar] [CrossRef]
- Hochgerner, H.; Zeisel, A.; Lönnerberg, P.; Linnarsson, S. Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat. Neurosci. 2018, 21, 290–299. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.R.; Manno, G.L.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lin, Y.; Ormerod, J.T.; Yang, P.; Yang, J.Y.H.; Lo, K.K. scDC: Single cell differential composition analysis. BMC Bioinform. 2019, 20 (Suppl. S19), 721. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.A.; Trapnell, C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L.; et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Avg_log2FC | Avg_log2FC Grn.KO | p-Value (Adjusted) |
|---|---|---|---|
| Csmd3 | −1.8672 | −1.148 | 1.73 × 10−95 |
| Cables1 | −0.4903 | −0.447 | 7.69 × 10−10 |
| Ptprm | −1.3229 | −0.399 | 3.16 × 10−40 |
| Plxdc2 | −0.6931 | −0.34 | 3.35 × 10−15 |
| Rapgef5 | −0.5133 | −0.313 | 6.18 × 10−15 |
| Tanc2 | −0.79899 | −0.252 | 1.61 × 10−26 |
| Bin1 | −0.33 | −0.094 | 6.49 × 10−8 |
| Ldlrad4 | −1.2606 | −0.022 | 1.19 × 10−31 |
| Fcrls | 0.7973 | 0.009 | 2.55 × 10−13 |
| Stab1 | 0.5704 | 0.269 | 9.19 × 10−4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Blanco, Á.; Sierra, C.; Tejido, C.; Dierssen, M. Beyond Quiescent and Active: Intermediate Microglial Transcriptomic States in a Mouse Model of Down Syndrome. Int. J. Mol. Sci. 2024, 25, 3289. https://doi.org/10.3390/ijms25063289
Fernández-Blanco Á, Sierra C, Tejido C, Dierssen M. Beyond Quiescent and Active: Intermediate Microglial Transcriptomic States in a Mouse Model of Down Syndrome. International Journal of Molecular Sciences. 2024; 25(6):3289. https://doi.org/10.3390/ijms25063289
Chicago/Turabian StyleFernández-Blanco, Álvaro, Cèsar Sierra, Clara Tejido, and Mara Dierssen. 2024. "Beyond Quiescent and Active: Intermediate Microglial Transcriptomic States in a Mouse Model of Down Syndrome" International Journal of Molecular Sciences 25, no. 6: 3289. https://doi.org/10.3390/ijms25063289