
The Role of the Endocrine System in the Regulation of Acid–Base Balance by the Kidney and the Progression of Chronic Kidney Disease
Abstract
:1. Introduction
2. Regulation of Acid–Base Balance
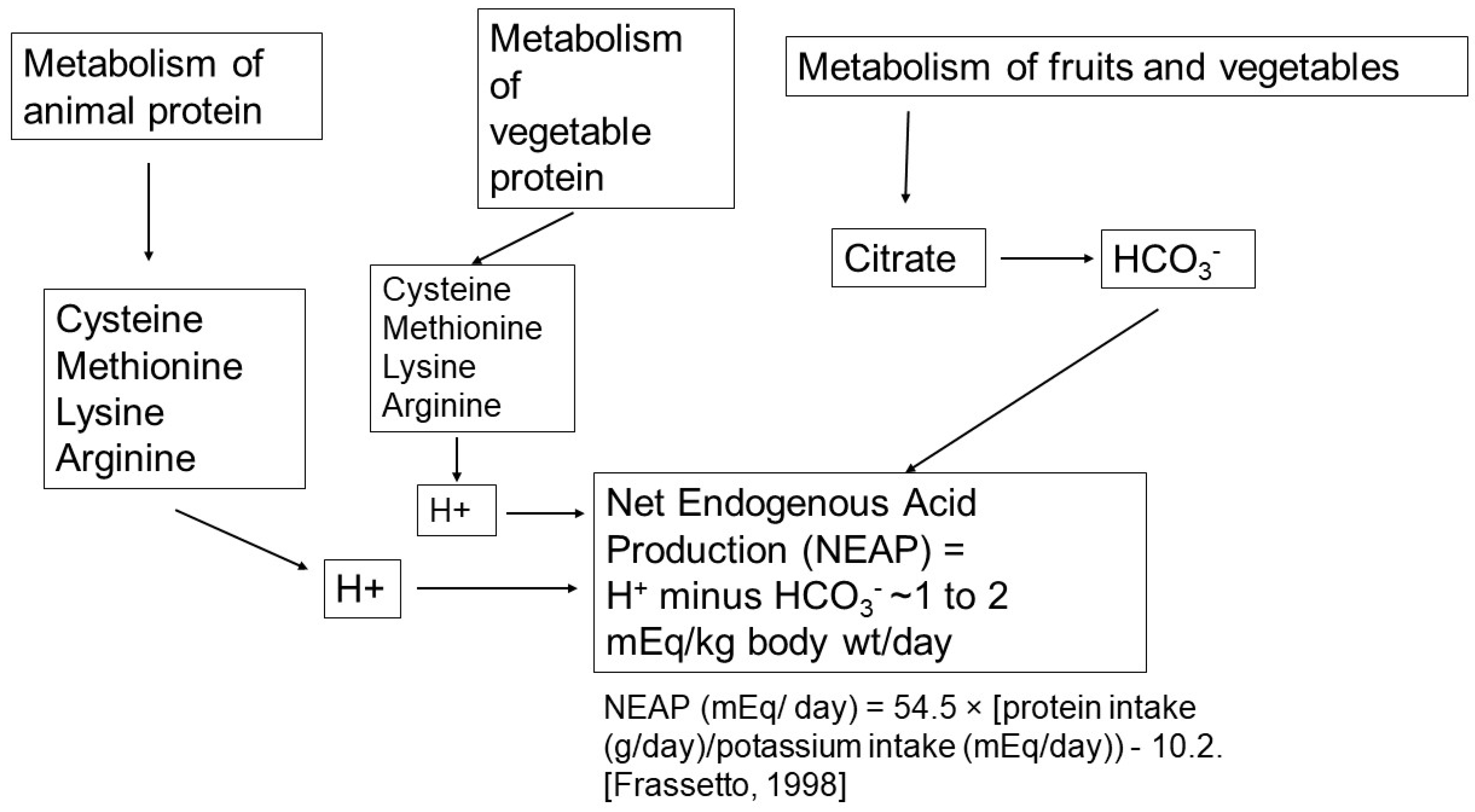
2.1. Net Acid Production
2.2. Buffering of Acid by Tissues
2.3. Bicarbonate Reabsorption and Generation by the Kidneys
3. Roles of Hormones in Acid-Balance and in Ckd Progression
3.1. Aldosterone
3.2. Angiotensin II
3.3. Endothelin
3.4. Parathyroid Hormone
3.5. Glucocorticoids
3.6. Insulin
3.7. Antidiuretic Hormone
3.8. Thyroid Hormone
3.9. Growth Hormone and Insulin-like Growth Factor 1
3.10. Effects of Acid pH Which May Contribute to CKD Progression in the Absence of Hormones
4. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Remer, T.; Manz, F. Estimation of the renal net acid excretion by adults consuming diets containing variable amounts of protein. Am. J. Clin. Nutr. 1994, 59, 1356–1361. [Google Scholar] [CrossRef]
- Lennon, E.J.; Lemann, J., Jr.; Litzow, J.R. The effects of diet and stool composition on the net external acid balance of normal subjects. J. Clin. Investig. 1966, 45, 1601–1607. [Google Scholar] [CrossRef]
- Toba, K.; Hosojima, M.; Kabasawa, H.; Kuwahara, S.; Murayama, T.; Yamamoto-Kabasawa, K.; Kaseda, R.; Wada, E.; Watanabe, R.; Tanabe, N.; et al. Higher estimated net endogenous acid production with lower intake of fruits and vegetables based on a dietary survey is associated with the progression of chronic kidney disease. BMC Nephrol. 2019, 20, 421. [Google Scholar] [CrossRef]
- Kurtz, I.; Maher, T.; Hulter, H.N.; Schambelan, M.; Sebastian, A. Effect of diet on plasma acid-base composition in normal humans. Kidney Int. 1983, 24, 670–680. [Google Scholar] [CrossRef]
- Frassetto, L.A.; Todd, K.M.; Morris, R.C., Jr.; Sebastian, A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am. J. Clin. Nutr. 1998, 68, 576–583. [Google Scholar] [CrossRef]
- Scialla, J.J.; Appel, L.J.; Astor, B.C.; Miller, E.R., 3rd; Beddhu, S.; Woodward, M.; Parekh, R.S.; Anderson, C.A. Estimated net endogenous acid production and serum bicarbonate in african americans with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.D.; Lemann, J., Jr.; Lennon, E.J.; Relman, A.S. Production, excretion, and net balance of fixed acid in patients with renal acidosis. J. Clin. Investig. 1965, 44, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Douyon, H.; Oh, M.S. A re-evaluation of the urinary parameters of acid production and excretion in patients with chronic renal acidosis. Kidney Int. 1995, 47, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Buquing, J.; Oh, M.S. Acid-base balance in chronic peritoneal dialysis patients. Kidney Int. 1995, 47, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Zia, M.; Mahmood, J.; Marcus, R.A.; Oh, M.S. Acid production in chronic hemodialysis patients. J. Am. Soc. Nephrol. 1998, 9, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Marangella, M.; Vitale, C.; Manganaro, M.; Cosseddu, D.; Martini, C.; Petrarulo, M.; Linari, F. Renal handling of citrate in chronic renal insufficiency. Nephron 1991, 57, 439–443. [Google Scholar] [CrossRef]
- Lisawat, P.; Gennari, F.J. Approach to the hemodialysis patient with an abnormal serum bicarbonate concentration. Am. J. Kidney Dis. 2014, 64, 151–155. [Google Scholar] [CrossRef]
- Goraya, N.; Munoz-Maldonado, Y.; Simoni, J.; Wesson, D.E. Treatment of chronic kidney disease-related metabolic acidosis with fruits and vegetables compared to nahco3 yields more and better overall health outcomes and at comparable five-year cost. J. Ren. Nutr. 2021, 31, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Del Canale, S.; Fiaccadori, E.; Coffrini, E.; Vitali, P.; Ronda, N.; Antonucci, C.; Arduini, U.; Guariglia, A. Uremic acidosis and intracellular buffering. Scand. J. Urol. Nephrol. 1986, 20, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Anderson, C.A. Dietary acid load: A novel nutritional target in chronic kidney disease? Adv. Chronic Kidney Dis. 2013, 20, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Lemann, J., Jr.; Bushinsky, D.A.; Hamm, L.L. Bone buffering of acid and base in humans. Am. J. Physiol. Renal. Physiol. 2003, 285, F811–F832. [Google Scholar] [CrossRef]
- Bushinsky, D.A.; Krieger, N.S. Effects of acid on bone. Kidney Int. 2022, 101, 1160–1170. [Google Scholar] [CrossRef]
- Frassetto, L.; Banerjee, T.; Powe, N.; Sebastian, A. Acid balance, dietary acid load, and bone effects-a controversial subject. Nutrients 2018, 10, 517. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, P.; Pluznick, J.L. Acid-base regulation in the renal proximal tubules: Using novel ph sensors to maintain homeostasis. Am. J. Physiol. Renal. Physiol. 2018, 315, F1187–F1190. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, B.M. The kidney and acid-base regulation. Adv. Physiol. Educ. 2009, 33, 275–281. [Google Scholar] [CrossRef]
- Schwartz, W.B.; Hall, P.W., 3rd; Hays, R.M.; Relman, A.S. On the mechanism of acidosis in chronic renal disease. J. Clin. Investig. 1959, 38, 39–52. [Google Scholar] [CrossRef]
- Arruda, J.A.; Nascimento, L.; Arevalo, G.; Baranowski, R.L.; Cubria, A.; Carrasquillo, T.; Westenfelder, C.; Kurtzman, N.A. Bicarbonate reabsorption in chronic renal failure studies in man and the rat. Pflug. Arch. 1978, 376, 193–199. [Google Scholar] [CrossRef]
- Relman, A.S. Renal acidosis and renal excretion of acid in health and disease. Adv. Intern. Med. 1964, 12, 295–347. [Google Scholar]
- Relman, A.S. The acidosis of renal disease. Am. J. Med. 1968, 44, 706–713. [Google Scholar] [CrossRef]
- Alpern, R.J.; Sakhaee, K. The clinical spectrum of chronic metabolic acidosis: Homeostatic mechanisms produce significant morbidity. Am. J. Kidney Dis. 1997, 29, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Madias, N.E. Eubicarbonatemic hydrogen ion retention and ckd progression. Kidney Med. 2021, 3, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. The continuum of acid stress. Clin. J. Am. Soc. Nephrol. 2021, 16, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Caso, G.; Garlick, P.J. Control of muscle protein kinetics by acid-base balance. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E. Metabolic acidosis stimulates protein metabolism in uremia. Miner. Electrol. Metab. 1996, 22, 62–65. [Google Scholar]
- Sebastian, A.; Harris, S.T.; Ottaway, J.H.; Todd, K.M.; Morris, R.C., Jr. Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N. Engl. J. Med. 1994, 330, 1776–1781. [Google Scholar] [CrossRef]
- Wesson, D.E.; Pruszynski, J.; Cai, W.; Simoni, J. Acid retention with reduced glomerular filtration rate increases urine biomarkers of kidney and bone injury. Kidney Int. 2016, 91, 914–927. [Google Scholar] [CrossRef]
- Wesson, D.E.; Buysse, J.M.; Bushinsky, D.A. Mechanisms of metabolic acidosis-induced kidney injury in chronic kidney disease. J. Am. Soc. Nephrol. 2020, 31, 469–482. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Schold, J.D.; Arrigain, S.; Jolly, S.E.; Wehbe, E.; Raina, R.; Simon, J.F.; Srinivas, T.R.; Jain, A.; Schreiber, M.J., Jr.; et al. Serum bicarbonate and mortality in stage 3 and stage 4 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2395–2402. [Google Scholar] [CrossRef]
- Perez, G.O.; Oster, J.R.; Vaamonde, C.A.; Katz, F.H. Effect of nh4cl on plasma aldosterone, cortisol and renin activity in supine man. J. Clin. Endocrinol. Metab. 1977, 45, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Schambelan, M.; Sebastian, A.; Katuna, B.A.; Arteaga, E. Adrenocortical hormone secretory response to chronic nh4cl-induced metabolic acidosis. Am. J. Physiol. 1987, 252, E454–E460. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Schulz, N.; Giebisch, G.; Geibel, J.P.; Wagner, C.A. Nongenomic stimulation of vacuolar h+-atpases in intercalated renal tubule cells by aldosterone. Proc. Natl. Acad. Sci. USA 2004, 101, 2636–2641. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Kampik, N.B.; Vedovelli, L.; Rothenberger, F.; Paunescu, T.G.; Stehberger, P.A.; Brown, D.; John, H.; Wagner, C.A. Aldosterone stimulates vacuolar h+-atpase activity in renal acid-secretory intercalated cells mainly via a protein kinase C-dependent pathway. Am. J. Physiol. Cell. Physiol. 2011, 301, C1251–C1261. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Izumi, Y.; Yasuoka, Y.; Nakagawa, T.; Ono, M.; Maruyama, K.; Matsuo, N.; Hiramatsu, A.; Inoue, H.; Nakayama, Y.; et al. Regulation of rhcg, an ammonia transporter, by aldosterone in the kidney. J. Endocrinol. 2021, 249, 95–112. [Google Scholar] [CrossRef]
- Bourgeois, S.; Bounoure, L.; Mouro-Chanteloup, I.; Colin, Y.; Brown, D.; Wagner, C.A. The ammonia transporter rhcg modulates urinary acidification by interacting with the vacuolar proton-atpases in renal intercalated cells. Kidney Int. 2018, 93, 390–402. [Google Scholar] [CrossRef]
- Henger, A.; Tutt, P.; Riesen, W.F.; Hulter, H.N.; Krapf, R. Acid-base and endocrine effects of aldosterone and angiotensin II inhibition in metabolic acidosis in human patients. J. Lab. Clin. Med. 2000, 136, 379–389. [Google Scholar] [CrossRef]
- Leavey, S.F.; Weitzel, W.F. Endocrine abnormalities in chronic renal failure. Endocrinol. Metab. Clin. N. Am. 2002, 31, 107–119. [Google Scholar] [CrossRef]
- Verma, A.; Vaidya, A.; Subudhi, S.; Waikar, S.S. Aldosterone in chronic kidney disease and renal outcomes. Eur. Heart J. 2022, 43, 3781–3791. [Google Scholar] [CrossRef]
- Ng, H.Y.; Chen, H.C.; Tsai, Y.C.; Yang, Y.K.; Lee, C.T. Activation of intrarenal renin-angiotensin system during metabolic acidosis. Am. J. Nephrol. 2011, 34, 55–63. [Google Scholar] [CrossRef]
- Geibel, J.; Giebisch, G.; Boron, W.F. Angiotensin ii stimulates both Na+-H+ exchange and Na+/HCO3− cotransport in the rabbit proximal tubule. Proc. Natl. Acad. Sci. USA 1990, 87, 7917–7920. [Google Scholar] [CrossRef]
- Eiam-Ong, S.; Hilden, S.A.; Johns, C.A.; Madias, N.E. Stimulation of basolateral Na+-HCO3− cotransporter by angiotensin ii in rabbit renal cortex. Am. J. Physiol. 1993, 265, F195–F203. [Google Scholar] [CrossRef]
- Nagami, G.T. Effect of angiotensin ii on ammonia production and secretion by mouse proximal tubules perfused in vitro. J. Clin. Investig. 1992, 89, 925–931. [Google Scholar] [CrossRef]
- Rothenberger, F.; Velic, A.; Stehberger, P.A.; Kovacikova, J.; Wagner, C.A. Angiotensin ii stimulates vacuolar h+-ATPase activity in renal acid-secretory intercalated cells from the outer medullary collecting duct. J. Am. Soc. Nephrol. 2007, 18, 2085–2093. [Google Scholar] [CrossRef]
- Mills, K.T.; Kobori, H.; Hamm, L.L.; Alper, A.B.; Khan, I.E.; Rahman, M.; Navar, L.G.; Liu, Y.; Browne, G.M.; Batuman, V.; et al. Increased urinary excretion of angiotensinogen is associated with risk of chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 3176–3181. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Endothelin role in kidney acidification. Semin. Nephrol. 2006, 26, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Tsuruoka, S.; Watanabe, S.; Purkerson, J.M.; Fujimura, A.; Schwartz, G.J. Endothelin and nitric oxide mediate adaptation of the cortical collecting duct to metabolic acidosis. Am. J. Physiol. Renal. Physiol. 2006, 291, F866–F873. [Google Scholar] [CrossRef] [PubMed]
- Laghmani, K.; Preisig, P.A.; Alpern, R.J. The role of endothelin in proximal tubule proton secretion and the adaptation to a chronic metabolic acidosis. J. Nephrol. 2002, 15 (Suppl. 5), S75–S87. [Google Scholar]
- Licht, C.; Laghmani, K.; Yanagisawa, M.; Preisig, P.A.; Alpern, R.J. An autocrine role for endothelin-1 in the regulation of proximal tubule NHE3. Kidney Int. 2004, 65, 1320–1326. [Google Scholar] [CrossRef]
- Wesson, D.E. Endogenous endothelins mediate increased acidification in remnant kidneys. J. Am. Soc. Nephrol. 2001, 12, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Kohan, D.E.; Barton, M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int. 2014, 86, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010, 78, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.M. Role of endothelin in chronic renal failure—Developments in renal involvement. Rheumatology 2006, 45 (Suppl. 3), iii36–iii38. [Google Scholar] [CrossRef] [PubMed]
- Hulter, H.N. Effects and interrelationships of PTH, Ca2+, vitamin D, and Pi in acid-base homeostasis. Am. J. Physiol. 1985, 248, F739–F752. [Google Scholar] [CrossRef]
- Campion, K.L.; McCormick, W.D.; Warwicker, J.; Khayat, M.E.; Atkinson-Dell, R.; Steward, M.C.; Delbridge, L.W.; Mun, H.C.; Conigrave, A.D.; Ward, D.T. Pathophysiologic changes in extracellular ph modulate parathyroid calcium-sensing receptor activity and secretion via a histidine-independent mechanism. J. Am. Soc. Nephrol. 2015, 26, 2163–2171. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Parving, H.H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (sonar): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Paillard, M.; Bichara, M. Peptide hormone effects on urinary acidification and acid-base balance: PTH, ADH, and glucagon. Am. J. Physiol. 1989, 256, F973–F985. [Google Scholar] [CrossRef]
- Lopez, I.; Aguilera-Tejero, E.; Estepa, J.C.; Rodriguez, M.; Felsenfeld, A.J. Role of acidosis-induced increases in calcium on pth secretion in acute metabolic and respiratory acidosis in the dog. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E780–E785. [Google Scholar] [CrossRef]
- Bichara, M.; Mercier, O.; Borensztein, P.; Paillard, M. Acute metabolic acidosis enhances circulating parathyroid hormone, which contributes to the renal response against acidosis in the rat. J. Clin. Investig. 1990, 86, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, P.; Isakova, T.; Asplin, J.; Hamm, L.; Dobre, M.; Rahman, M.; Sharma, K.; Leonard, M.; Miller, E., 3rd; Jaar, B.; et al. Acid load and phosphorus homeostasis in ckd. Am. J. Kidney Dis. 2017, 70, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Evans, M.; Soro, M.; Barany, P.; Carrero, J.J. Secondary hyperparathyroidism and adverse health outcomes in adults with chronic kidney disease. Clin. Kidney J. 2021, 14, 2213–2220. [Google Scholar] [CrossRef] [PubMed]
- Hulter, H.N.; Licht, J.H.; Bonner, E.L., Jr.; Glynn, R.D.; Sebastian, A. Effects of glucocorticoid steroids on renal and systemic acid-base metabolism. Am. J. Physiol. 1980, 239, F30–F43. [Google Scholar] [CrossRef] [PubMed]
- Perez, G.O.; Oster, J.R.; Katz, F.H.; Vaamonde, C.A. The effect of acute metabolic acidosis on plasma cortisol, renin activity and aldosterone. Horm. Res. 1979, 11, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, J.; Cujdik, T.; Sacktor, B. Na+-H+ exchange activity in renal brush border membrane vesicles in response to metabolic acidosis: The role of glucocorticoids. Proc. Natl. Acad. Sci. USA 1984, 81, 630–634. [Google Scholar] [CrossRef]
- Baum, M.; Moe, O.W.; Gentry, D.L.; Alpern, R.J. Effect of glucocorticoids on renal cortical NHE-3 and NHE-1 mRNA. Am. J. Physiol. 1994, 267, F437–F442. [Google Scholar] [CrossRef]
- Welbourne, T.C. Glucocorticoid and acid-base homeostasis: Effects on glutamine metabolism and transport. Am. J. Kidney Dis. 1989, 14, 293–297. [Google Scholar] [CrossRef]
- Welbourne, T.C. Role of glucocorticoids in regulating interorgan glutamine flow during chronic metabolic acidosis. Metabolism 1988, 37, 520–525. [Google Scholar] [CrossRef]
- Tizianello, A.; De Ferrari, G.; Garibotto, G.; Gurreri, G.; Robaudo, C. Renal metabolism of amino acids and ammonia in subjects with normal renal function and in patients with chronic renal insufficiency. J. Clin. Investig. 1980, 65, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Sagmeister, M.S.; Harper, L.; Hardy, R.S. Cortisol excess in chronic kidney disease—A review of changes and impact on mortality. Front. Endocrinol. 2022, 13, 1075809. [Google Scholar] [CrossRef] [PubMed]
- Mak, R.H. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998, 54, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Zhang, L.; Mitch, W.E. Molecular mechanisms of insulin resistance in chronic kidney disease. Kidney Int. 2015, 88, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Renal. Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.W.; Zhang, X.Y.; Tian, J.; Correale, J.D.; Xi, X.P.; Yang, D.; Graf, K.; Law, R.E.; Hsueh, W.A. Insulin and angiotensin ii are additive in stimulating tgf-beta 1 and matrix mrnas in mesangial cells. Kidney Int. 1996, 50, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Rocchini, A.P.; Katch, V.; Kveselis, D.; Moorehead, C.; Martin, M.; Lampman, R.; Gregory, M. Insulin and renal sodium retention in obese adolescents. Hypertension 1989, 14, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Catalano, C.; Muscelli, E.; Quinones Galvan, A.; Baldi, S.; Masoni, A.; Gibb, I.; Torffvit, O.; Seghieri, G.; Ferrannini, E. Effect of insulin on systemic and renal handling of albumin in nondiabetic and niddm subjects. Diabetes 1997, 46, 868–875. [Google Scholar] [CrossRef]
- Conti, F.G.; Striker, L.J.; Lesniak, M.A.; MacKay, K.; Roth, J.; Striker, G.E. Studies on binding and mitogenic effect of insulin and insulin-like growth factor i in glomerular mesangial cells. Endocrinology 1988, 122, 2788–2795. [Google Scholar] [CrossRef]
- Mohebbi, N.; Kovacikova, J.; Nowik, M.; Wagner, C.A. Thyroid hormone deficiency alters expression of acid-base transporters in rat kidney. Am. J. Physiol. Renal. Physiol. 2007, 293, F416–F427. [Google Scholar] [CrossRef]
- Bargagli, M.; Dhayat, N.A.; Anderegg, M.; Semmo, M.; Huynh-Do, U.; Vogt, B.; Ferraro, P.M.; Fuster, D.G. Urinary lithogenic risk profile in adpkd patients treated with tolvaptan. Clin. J. Am. Soc. Nephrol. 2020, 15, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Oster, J.R.; Michael, U.F.; Perez, G.O.; Sonneborn, R.E.; Vaamonde, C.A. Renal acidification in hypothyroid man. Clin. Nephrol. 1976, 6, 398–403. [Google Scholar] [PubMed]
- Michael, U.F.; Chavez, R.; Cookson, S.L.; Vaamonde, C.A. Impaired urinary acidification in the hypothyroid rat. Pflug. Arch. 1976, 361, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Michael, U.F.; Kelley, J.; Meeks, L.A.; Vaamonde, C.A. Renal acidification in the hypothyroid rat. Evaluation by urinary CO2 tension. Can. J. Physiol. Pharmacol. 1981, 59, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Brungger, M.; Hulter, H.N.; Krapf, R. Effect of chronic metabolic acidosis on thyroid hormone homeostasis in humans. Am. J. Physiol. 1997, 272, F648–F653. [Google Scholar] [CrossRef]
- Narasaki, Y.; Sohn, P.; Rhee, C.M. The interplay between thyroid dysfunction and kidney disease. Semin. Nephrol. 2021, 41, 133–143. [Google Scholar] [CrossRef]
- Parry-Billings, M.; Dimitriadis, G.D.; Leighton, B.; Bond, J.; Bevan, S.J.; Opara, E.; Newsholme, E.A. Effects of hyperthyroidism and hypothyroidism on glutamine metabolism by skeletal muscle of the rat. Biochem. J. 1990, 272, 319–322. [Google Scholar] [CrossRef]
- Decaux, G.; Musch, W. Clinical laboratory evaluation of the syndrome of inappropriate secretion of antidiuretic hormone. Clin. J. Am. Soc. Nephrol. 2008, 3, 1175–1184. [Google Scholar] [CrossRef]
- Cohen, J.J.; Hulter, H.N.; Smithline, N.; Melby, J.C.; Schwartz, W.B. The critical role of the adrenal gland in the renal regulation of acid-base equilibrium during chronic hypotonic expansion. Evidence that chronic hyponatremia is a potent stimulus to aldosterone secretion. J. Clin. Investig. 1976, 58, 1201–1208. [Google Scholar] [CrossRef]
- Walsh, C.H.; Baylis, P.H.; Malins, J.M. Plasma arginine vasopressin in diabetic ketoacidosis. Diabetologia 1979, 16, 93–96. [Google Scholar] [CrossRef]
- Kakeshita, K.; Koike, T.; Imamura, T.; Fujioka, H.; Yamazaki, H.; Kinugawa, K. Altered arginine vasopressin-cyclic amp-aquaporin 2 pathway in patients with chronic kidney disease. Clin. Exp. Nephrol. 2022, 26, 788–796. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef]
- Corvilain, J.; Abramow, M.; Bergans, A. Some effects of human growth hormone on renal hemodynamics and on tubular phosphate transport in man. J. Clin. Investig. 1962, 41, 1230–1235. [Google Scholar] [CrossRef]
- Chobanian, M.C.; Julin, C.M.; Molteni, K.H.; Brazy, P.C. Growth hormone regulates ammoniagenesis in canine renal proximal tubule segments. Am. J. Physiol. 1992, 262, F878–F884. [Google Scholar] [CrossRef] [PubMed]
- Sicuro, A.; Mahlbacher, K.; Hulter, H.N.; Krapf, R. Effect of growth hormone on renal and systemic acid-base homeostasis in humans. Am. J. Physiol. 1998, 274, F650–F657. [Google Scholar] [CrossRef] [PubMed]
- Welbourne, T.C.; Cronin, M.J. Growth hormone accelerates tubular acid secretion. Am. J. Physiol. 1991, 260, R1036–R1042. [Google Scholar] [CrossRef] [PubMed]
- Jehle, S.; Hulter, H.N.; Krapf, R. On the mechanism of growth hormone-induced stimulation of renal acidification in humans: Effect of dietary NaCL. Clin. Sci. 2000, 99, 47–56. [Google Scholar] [CrossRef]
- Quigley, R.; Baum, M. Effects of growth hormone and insulin-like growth factor i on rabbit proximal convoluted tubule transport. J. Clin. Investig. 1991, 88, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, S.; Kaskel, F. Growth hormone axis in chronic kidney disease. Pediatr. Nephrol. 2008, 23, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.B.; Hansen, V.A.; Hammerman, M.R. Effects of growth hormone and igf-i on renal function in rats with normal and reduced renal mass. Am. J. Physiol. 1990, 259, F747–F751. [Google Scholar] [CrossRef]
- Auriemma, R.S.; Galdiero, M.; De Martino, M.C.; De Leo, M.; Grasso, L.F.; Vitale, P.; Cozzolino, A.; Lombardi, G.; Colao, A.; Pivonello, R. The kidney in acromegaly: Renal structure and function in patients with acromegaly during active disease and 1 year after disease remission. Eur. J. Endocrinol. 2010, 162, 1035–1042. [Google Scholar] [CrossRef]
- Takai, M.; Izumino, K.; Oda, Y.; Terada, Y.; Inoue, H.; Takata, M. Focal segmental glomerulosclerosis associated with acromegaly. Clin. Nephrol. 2001, 56, 75–77. [Google Scholar]
- Schambelan, M.; Sebastian, A.; Biglieri, E.G. Prevalence, pathogenesis, and functional significance of aldosterone deficiency in hyperkalemic patients with chronic renal insufficiency. Kidney Int. 1980, 17, 89–101. [Google Scholar] [CrossRef]
- Szylman, P.; Better, O.S.; Chaimowitz, C.; Rosler, A. Role of hyperkalemia in the metabolic acidosis of isolated hypoaldosteronism. N. Engl. J. Med. 1976, 294, 361–365. [Google Scholar] [CrossRef]
- Harris, A.N.; Grimm, P.R.; Lee, H.W.; Delpire, E.; Fang, L.; Verlander, J.W.; Welling, P.A.; Weiner, I.D. Mechanism of hyperkalemia-induced metabolic acidosis. J. Am. Soc. Nephrol. 2018, 29, 1411–1425. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J.; Broglio, K.; Sheather, S. Acid retention accompanies reduced gfr in humans and increases plasma levels of endothelin and aldosterone. Am. J. Physiol. Renal. Physiol. 2011, 300, F830–F837. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney Int. 1980, 17, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Nagami, G.T. Effect of luminal angiotensin ii on ammonia production and secretion by mouse proximal tubules. Am. J. Physiol. 1995, 269, F86–F92. [Google Scholar] [CrossRef] [PubMed]
- Nagami, G.T. Role of angiotensin ii in the enhancement of ammonia production and secretion by the proximal tubule in metabolic acidosis. Am. J. Physiol. Renal. Physiol. 2008, 294, F874–F880. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L.; Zhang, Y.; Ying, J.; Greene, T. Prevalence of and risk factors for reduced serum bicarbonate in chronic kidney disease. Nephrology 2014, 19, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Yanagisawa, M. Endothelin: 30 years from discovery to therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef]
- Bichara, M.; Mercier, O.; Paillard, M.; Leviel, F. Effects of parathyroid hormone on urinary acidification. Am. J. Physiol. 1986, 251, F444–F453. [Google Scholar] [CrossRef]
- Arruda, J.A.; Alla, V.; Rubinstein, H.; Cruz-Soto, M.; Sabatini, S.; Batlle, D.C.; Kurtzman, N.A. Metabolic and hormonal factors influencing extrarenal buffering of an acute acid load. Miner. Electrolyte Metab. 1982, 8, 36–43. [Google Scholar] [PubMed]
- Bozic, M.; Diaz-Tocados, J.M.; Bermudez-Lopez, M.; Forne, C.; Martinez, C.; Fernandez, E.; Valdivielso, J.M. Independent effects of secondary hyperparathyroidism and hyperphosphataemia on chronic kidney disease progression and cardiovascular events: An analysis from the nefrona cohort. Nephrol. Dial. Transplant. 2022, 37, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Malnic, G.; Ansaldo, M.; Lantos, C.P.; Damasco, M.C. Regulation of nephron acidification by corticosteroids. Braz. J. Med. Biol. Res. 1997, 30, 479–486. [Google Scholar] [CrossRef]
- Field, M.J.; Stanton, B.A.; Giebisch, G.H. Differential acute effects of aldosterone, dexamethasone, and hyperkalemia on distal tubular potassium secretion in the rat kidney. J. Clin. Investig. 1984, 74, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Seldin, D.W.; Coleman, A.J.; Carter, N.W.; Rector, F.C.J. The effect of Na2SO4 on urinary acidification in chronic renal disease. J. Lab. Clin. Med. 1967, 69, 893–903. [Google Scholar] [PubMed]
- May, R.C.; Kelly, R.A.; Mitch, W.E. Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism. J. Clin. Investig. 1986, 77, 614–621. [Google Scholar] [CrossRef]
- Walser, M.; Ward, L. Progression of chronic renal failure is related to glucocorticoid production. Kidney Int. 1988, 34, 859–866. [Google Scholar] [CrossRef]
- Takahashi, E.; Onda, K.; Hirano, T.; Oka, K.; Maruoka, N.; Tsuyuguchi, M.; Matsumura, Y.; Niitsuma, T.; Hayashi, T. Expression of c-fos, rather than c-jun or glucocorticoid-receptor mRNA, correlates with decreased glucocorticoid response of peripheral blood mononuclear cells in asthma. Int. Immunopharmacol. 2002, 2, 1419–1427. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Cooke, C.R.; Andres, R.; Faloona, G.R.; Davis, P.J. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J. Clin. Investig. 1975, 55, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Brands, M.W. Role of insulin-mediated antinatriuresis in sodium homeostasis and hypertension. Hypertension 2018, 72, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Arruda, J.A.; Kumar, S.; Wilson, A.; Sehy, J.T.; Roseman, M.K.; Nascimento, L.; Kurtzman, N.A. Distal acidification defect caused by pharmacological doses of insulin. J. Lab. Clin. Med. 1980, 95, 440–449. [Google Scholar] [PubMed]
- Gerich, J.E.; Meyer, C.; Stumvoll, M.W. Hormonal control of renal and systemic glutamine metabolism. J. Nutr. 2000, 130, 995S–1001S. [Google Scholar] [CrossRef]
- Bellasi, A.; Di Micco, L.; Santoro, D.; Marzocco, S.; De Simone, E.; Cozzolino, M.; Di Lullo, L.; Guastaferro, P.; Di Iorio, B.; UBI Study Investigators. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Bichara, M.; Mercier, O.; Houillier, P.; Paillard, M.; Leviel, F. Effects of antidiuretic hormone on urinary acidification and on tubular handling of bicarbonate in the rat. J. Clin. Investig. 1987, 80, 621–630. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Bargagli, M.; Vetsch, A.; Anderegg, M.A.; Dhayat, N.A.; Huynh-Do, U.; Faller, N.; Vogt, B.; Ferraro, P.M.; Fuster, D.G. Tolvaptan treatment is associated with altered mineral metabolism parameters and increased bone mineral density in adpkd patients. Nephrol. Dial. Transplant. 2023, 38, 1645–1654. [Google Scholar] [CrossRef]
- Wiederkehr, M.R.; Kalogiros, J.; Krapf, R. Correction of metabolic acidosis improves thyroid and growth hormone axes in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 1190–1197. [Google Scholar] [CrossRef]
- Brungger, M.; Hulter, H.N.; Krapf, R. Effect of chronic metabolic acidosis on the growth hormone igf-1 endocrine axis: New cause of growth hormone insensitivity in humans. Kidney Int. 1997, 51, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Brosius, F.C., 3rd; Menon, R.K. The glomerular podocyte as a target of growth hormone action: Implications for the pathogenesis of diabetic nephropathy. Curr. Diabetes Rev. 2011, 7, 50–55. [Google Scholar] [CrossRef]
- Wuhl, E.; Haffner, D.; Gretz, N.; Offner, G.; van’t Hoff, W.G.; Broyer, M.; Mehls, O. Treatment with recombinant human growth hormone in short children with nephropathic cystinosis: No evidence for increased deterioration rate of renal function. The european study group on growth hormone treatment in short children with nephropathic cystinosis. Pediatr. Res. 1998, 43, 484–488. [Google Scholar] [PubMed]
- Hodson, E.M.; Willis, N.S.; Craig, J.C. Growth hormone for children with chronic kidney disease. Cochrane Database Syst. Rev. 2012, 2012, CD003264. [Google Scholar] [CrossRef]
- Mehls, O.; Lindberg, A.; Haffner, D.; Schaefer, F.; Wuhl, E.; German, K.B.; Group, E.T. Long-term growth hormone treatment in short children with ckd does not accelerate decline of renal function: Results from the kigs registry and escape trial. Pediatr. Nephrol. 2015, 30, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Song, M.; Li, J. Lactic and hydrochloric acids induce different patterns of inflammatory response in lps-stimulated raw 264.7 cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R686–R692. [Google Scholar] [CrossRef]
- de Nadai, T.R.; de Nadai, M.N.; Albuquerque, A.A.; de Carvalho, M.T.; Celotto, A.C.; Evora, P.R. Metabolic acidosis treatment as part of a strategy to curb inflammation. Int. J. Inflam. 2013, 2013, 601424. [Google Scholar] [CrossRef] [PubMed]
- Bugarski, M.; Ghazi, S.; Polesel, M.; Martins, J.R.; Hall, A.M. Changes in nad and lipid metabolism drive acidosis-induced acute kidney injury. J. Am. Soc. Nephrol. 2021, 32, 342–356. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Hormone | Effect of Acidosis on Serum Level or Action | Effect on Acid Base Balance | Role Acid–Base Balance in CKD | Effect on Progression of CKD |
|---|---|---|---|---|
| Aldosterone | Increased levels with metabolic acidosis [34,35] | Increases H+ secretion in collecting duct and increases net acid excretion [36,37,38] | Increased levels may help increase kidney acid excretion. Decreased levels may worsen acidosis [39,40] | Increased action associated with worsening kidney function. Blocking action slows progression [41,42] |
| Angiotensin II | Increased levels with metabolic acidosis [43] | Stimulates proximal tubule ammonia production and collecting duct acid excretion [44,45,46,47] | May help to preserve kidney acid excretion as kidney function declines [32] | A contributory cause of worsening kidney function. Blocking actions slows progression [32,42,48] |
| Endothelin | Increased with metabolic acidosis [49,50] | Increased net acid excretion by stimulating H+ secretion in proximal tubules and collecting duct [49,50,51,52,53] | May enhance acid excretion as kidney function declines [53,54] | Proinflammatory and profibrotic actions worsen kidney function [54,55,56] |
| PTH | Increased levels in some studies. [57,58] | Increased net acid excretion. Increased bone buffering capacity [17,58,59,60,61,62] | Increased levels in secondary hyperparathyroidism [63] | Association of secondary hyperparathyroidism with worsening CKD, but causal relationship is unclear [63,64] |
| Glucocorticoids | Increased levels [65] | Increased glutamine delivery and kidney ammonia production. increased proximal tubule acid secretion [66,67,68,69] | Higher levels of glucocorticoids in CKD are not associated with enhanced glutamine delivery. Unclear whether there is resistance to action [70] | Association of high glucocorticoids with progression but unclear whether the high glucocorticoid levels cause worsening of CKD [71,72] |
| Insulin | Increased levels due to insulin resistance [73,74] | High levels may reduce H+ secretion by distal nephron [75] | Effect on acid–base balance unclear. Correction of acidosis improves insulin sensitivity in CKD [76] | Hyperinsulinemia in the setting of insulin resistance may result in impaired kidney hemodynamics and progression of CKD [77,78,79] |
| Thyroid hormone | Reduced free T3 levels and increased TSH [80] | Hypothyroidism impedes maximal response to metabolic acidosis with reduced distal acidification and reduced tissue buffering [81,82,83,84,85] | Reduced T3 due to reduced conversion of T4. May improve with correction of acidosis [86] | Hypothyroidism in CKD is associated with poor outcomes (mortality, cardio-vascular disease, functional status, body composition) [87] |
| Antidiuretic hormone | Variable | May increase acid secretion in collecting duct leading to normal bicarbonate in SIADH [88,89] | Role in acid–base regulation in CKD is unclear as AVP signaling pathway may be disturbed [90] | Blocking action in patients with more rapidly progressive ADPKD can slow progression. [91,92] |
| Growth hormone (GH) and IGF-1 | GH may be increased while IGF-1 levels may be decreased [93,94] | Reduced GH and IGF-1 activity may impair tissue buffering by loss of muscle and bone mass. Direct effects of GH on tubular acid secretion [93,95,96,97,98] | In severe CKD, GH levels are normal or increased but there is resistance to GH action accompanied by reduced IGF-1 levels. Unknown if GH receptors are reduced in renal tissue [98] | High GH levels may be associated with progressive kidney disease under certain conditions through adverse effects on podocytes [99,100,101,102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagami, G.T.; Kraut, J.A. The Role of the Endocrine System in the Regulation of Acid–Base Balance by the Kidney and the Progression of Chronic Kidney Disease. Int. J. Mol. Sci. 2024, 25, 2420. https://doi.org/10.3390/ijms25042420
Nagami GT, Kraut JA. The Role of the Endocrine System in the Regulation of Acid–Base Balance by the Kidney and the Progression of Chronic Kidney Disease. International Journal of Molecular Sciences. 2024; 25(4):2420. https://doi.org/10.3390/ijms25042420
Chicago/Turabian StyleNagami, Glenn T., and Jeffrey A. Kraut. 2024. "The Role of the Endocrine System in the Regulation of Acid–Base Balance by the Kidney and the Progression of Chronic Kidney Disease" International Journal of Molecular Sciences 25, no. 4: 2420. https://doi.org/10.3390/ijms25042420