
Primary Human M2 Macrophage Subtypes Are Distinguishable by Aqueous Metabolite Profiles
 ,
, 
Abstract
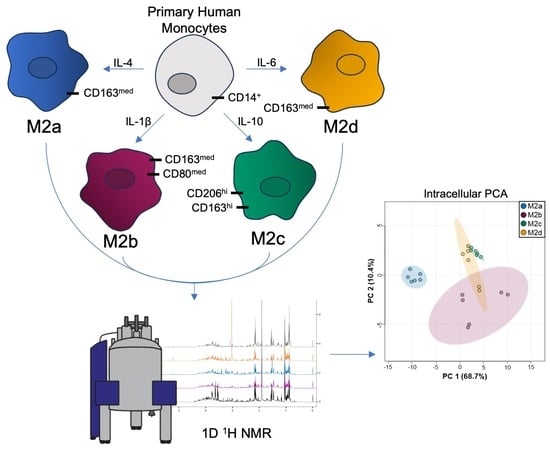
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Quantitative Metabolic Profiles Reveal M2a MΦs Are Remarkably Distinct from Other M2 MΦ Subtypes
2.2. Unique Intracellular Metabolite Signatures Reveal Significant Metabolic Traits Associated with M2b, M2c, and M2d MΦ Subtypes
2.3. Variability in Extracellular Metabolite Profiles of M2b, M2c, and M2d MΦ Subtypes Does Not Occlude the Ability to Separate These M2 MΦ Subtypes Based upon Characteristic Extracellular Water Soluble Metabolite Patterns
2.4. Analysis of M2b, M2c, and M2d Intra- and Extracellular Metabolomics Datasets Identified Shared Metabolites as Significant Discriminators of M2 MΦ Subtypes
3. Discussion
3.1. Distinct Elevation of Myo-Inositol in M2a MΦs Is Chiefly Attributable to Its Specialized Function
3.2. Metabolites Involved in Glycolysis, the TCA Cycle, and OXPHOS Differentiate M2a MΦs from M2b, M2c, and M2d MΦ Subtypes, Suggesting That Metabolome Differences Report on M2 vs. M1-like MΦ Phenotypic Distinctions
3.3. Elevated Levels of Amino Acids in M2b, M2c, and M2d MΦs Relative to M2a MΦs
3.4. Evidence of PPP Activity and Antioxidant Glutathione Metabolism Intersecting with Glycolysis, Glycogenolysis, and Amino Acid Metabolism
3.5. Characteristic Metabolite Profiles Further Differentiate the Distinct Metabolic States of M2b, M2c and M2d MΦs Subtypes
3.5.1. Lactate and TCA Cycle Intermediates Suggest That M2b MΦs Most Closely Adopt an M1 Phenotype Compared with M2c and M2d MΦs
3.5.2. Creatine Metabolism Differentiates M2b, M2c, and M2d MΦ Subtypes and Mediates Phagocytosis
3.5.3. Phospholipid Synthesis Is Impaired in M2c and M2d MΦ Subtypes
4. Materials and Methods
4.1. Isolation of Human Monocytes
4.2. Differentiation and Activation of Human Monocyte-Derived MΦs
4.3. Antibodies and Fluorescence-Activated Cell Sorting (FACS) Analysis
4.4. Extraction of Intra- and Extracellular Polar Metabolites for 1H NMR Analysis
4.5. 1H NMR Spectroscopy
4.6. NMR Data Processing, Metabolite Identification, and Quantitation
4.7. Determination of Intracellular Protein Concentrations and Sample Biomass Normalization
4.8. Univariate and Multivariate Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, G.; Schaible, U.E. Macrophage Defense Mechanisms against Intracellular Bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Plüddemann, A.; Mukhopadhyay, S.; Gordon, S. Innate Immunity to Intracellular Pathogens: Macrophage Receptors and Responses to Microbial Entry. Immunol. Rev. 2011, 240, 11–24. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage Plasticity, Polarization, and Function in Health and Disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 Polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Shrivastava, R.; Shukla, N. Attributes of Alternatively Activated (M2) Macrophages. Life Sci. 2019, 224, 222–231. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage Activation and Polarization. Front. Biosci. J. Virtual Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage Phenotypes in Atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, S.; Wu, H.; Rong, X.; Guo, J. M2b Macrophage Polarization and Its Roles in Diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef]
- Mosser, D.M. The Many Faces of Macrophage Activation. J. Leukoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism Governs Dendritic Cell and Macrophage Function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the Interface between Macrophages and Pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Benmoussa, K.; Garaude, J.; Acín-Pérez, R. How Mitochondrial Metabolism Contributes to Macrophage Phenotype and Functions. J. Mol. Biol. 2018, 430, 3906–3921. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.L.; Schiller, S.M.; Keegan, W.J.; Ammons, M.C.B.; Eilers, B.; Tripet, B.; Copié, V. Quantitative 1H NMR Metabolomics Reveal Distinct Metabolic Adaptations in Human Macrophages Following Differential Activation. Metabolites 2019, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-Check: Validation of Diagnostic Statistics for PLS-DA Models in Metabolomics Studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.T.; Hanjani, N.A.; Movahed, E.; Doroudian, M. The Role of Macrophage Subtypes and Exosomes in Immunomodulation. Cell. Mol. Biol. Lett. 2022, 27, 83. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, M.H.; Abdelwahab, S.F.; Wan, J.; Cai, W.; Huixuan, W.; Jianjun, C.; Kumar, K.D.; Vasudevan, A.; Sadek, A.; Su, Z.; et al. Alternatively Activated Macrophages; a Double-Edged Sword in Allergic Asthma. J. Transl. Med. 2020, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.B.; Lawton, T.M.; Smith, H.L.; Garret, J.; Doucette, M.M.; Ammons, M.C.B. Use of Integrated Metabolomics, Transcriptomics, and Signal Protein Profile to Characterize the Effector Function and Associated Metabotype of Polarized Macrophage Phenotypes. J. Leukoc. Biol. 2022, 111, 667–693. Available online: https://jlb.onlinelibrary.wiley.com/doi/10.1002/JLB.6A1120-744R (accessed on 28 October 2021). [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic Reprogramming in Macrophage Responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Kieler, M.; Hofmann, M.; Schabbauer, G. More than Just Protein Building Blocks: How Amino Acids and Related Metabolic Pathways Fuel Macrophage Polarization. FEBS J. 2021, 288, 3694–3714. [Google Scholar] [CrossRef]
- Everts, B. Metabolomics in Immunology Research. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1730, pp. 29–42. ISBN 978-1-4939-7591-4. [Google Scholar]
- Sowers, M.L.; Tang, H.; Singh, V.K.; Khan, A.; Mishra, A.; Restrepo, B.I.; Jagannath, C.; Zhang, K. Multi-OMICs Analysis Reveals Metabolic and Epigenetic Changes Associated with Macrophage Polarization. J. Biol. Chem. 2022, 298, 102418. [Google Scholar] [CrossRef]
- LI, P.; MA, C.; LI, J.; YOU, S.; DANG, L.; WU, J.; HAO, Z.; LI, J.; ZHI, Y.; CHEN, L.; et al. Proteomic Characterization of Four Subtypes of M2 Macrophages Derived from Human THP-1 Cells. J. Zhejiang Univ. Sci. B 2022, 23, 407–422. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Biswas, N.; Mahajan, S.P.; Madeshiya, A.K.; Khanna, S.; Sen, C.K.; Roy, S. Myo-Inositol in Fermented Sugar Matrix Improves Human Macrophage Function. Mol. Nutr. Food Res. 2022, 66, e2100852. [Google Scholar] [CrossRef]
- Codo, A.C.; de Aquino Penteado, L.; de Medeiros, A.I.; de Moraes-Vieira, P.M.M. Metabolic Profile of Innate Immune Cells. In Essential Aspects of Immunometabolism in Health and Disease; Camara, N.O.S., Alves-Filho, J.C., de Moraes-Vieira, P.M.M., Andrade-Oliveira, V., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 83–114. ISBN 978-3-030-86684-6. [Google Scholar]
- Ma, J.; Wei, K.; Liu, J.; Tang, K.; Zhang, H.; Zhu, L.; Chen, J.; Li, F.; Xu, P.; Chen, J.; et al. Glycogen Metabolism Regulates Macrophage-Mediated Acute Inflammatory Responses. Nat. Commun. 2020, 11, 1769. [Google Scholar] [CrossRef] [PubMed]
- ZHENG, J. Energy Metabolism of Cancer: Glycolysis versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Diskin, C.; Pålsson-McDermott, E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018, 9, 270. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.W.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.-C.; Wang, S.-Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Bredahl, E.C.; Eckerson, J.M.; Tracy, S.M.; McDonald, T.L.; Drescher, K.M. The Role of Creatine in the Development and Activation of Immune Responses. Nutrients 2021, 13, 751. [Google Scholar] [CrossRef]
- Kazak, L.; Cohen, P. Creatine Metabolism: Energy Homeostasis, Immunity and Cancer Biology. Nat. Rev. Endocrinol. 2020, 16, 421–436. [Google Scholar] [CrossRef]
- Darabedian, N.; Ji, W.; Fan, M.; Lin, S.; Seo, H.-S.; Vinogradova, E.V.; Yaron, T.M.; Mills, E.L.; Xiao, H.; Senkane, K.; et al. Depletion of Creatine Phosphagen Energetics with a Covalent Creatine Kinase Inhibitor. Nat. Chem. Biol. 2023, 19, 815–824. [Google Scholar] [CrossRef]
- Loike, J.D.; Kozler, V.F.; Silverstein, S.C. Increased ATP and Creatine Phosphate Turnover in Phagocytosing Mouse Peritoneal Macrophages. J. Biol. Chem. 1979, 254, 9558–9564. [Google Scholar] [CrossRef]
- Purcu, D.U.; Korkmaz, A.; Gunalp, S.; Helvaci, D.G.; Erdal, Y.; Dogan, Y.; Suner, A.; Wingender, G.; Sag, D. Effect of Stimulation Time on the Expression of Human Macrophage Polarization Markers. PLoS ONE 2022, 17, e0265196. [Google Scholar] [CrossRef]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef]
- Kaplanski, G. Interleukin--18: Biological Properties and Role in Disease Pathogenesis. Immunol. Rev. 2018, 281, 138–153. [Google Scholar] [CrossRef]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.-P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-Associated Leukemia Inhibitory Factor and IL-6 Skew Monocyte Differentiation into Tumor-Associated Macrophage-like Cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef]
- Fuchs, A.L.; Miller, I.R.; Schiller, S.M.; Ammons, M.C.B.; Eilers, B.; Tripet, B.; Copié, V. Pseudomonas Aeruginosa Planktonic- and Biofilm-Conditioned Media Elicit Discrete Metabolic Responses in Human Macrophages. Cells 2020, 9, 2260. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- O’Shea-Stone, G.; Lambert, R.; Tripet, B.; Berardinelli, J.; Thomson, J.; Copié, V.; Garrott, R. 1H NMR Based Metabolic Profiling Distinguishes the Differential Impact of Capture Techniques on Wild Bighorn Sheep. Sci. Rep. 2021, 11, 11308. [Google Scholar] [CrossRef]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Fabriek, B.O.; van Bruggen, R.; Deng, D.M.; Ligtenberg, A.J.M.; Nazmi, K.; Schornagel, K.; Vloet, R.P.M.; Dijkstra, C.D.; van den Berg, T.K. The Macrophage Scavenger Receptor CD163 Functions as an Innate Immune Sensor for Bacteria. Blood 2009, 113, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Katakura, T.; Miyazaki, M.; Kobayashi, M.; Herndon, D.N.; Suzuki, F. CCL17 and IL-10 as Effectors That Enable Alternatively Activated Macrophages to Inhibit the Generation of Classically Activated Macrophages1. J. Immunol. 2004, 172, 1407–1413. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, A.L.; Costello, S.M.; Schiller, S.M.; Tripet, B.P.; Copié, V. Primary Human M2 Macrophage Subtypes Are Distinguishable by Aqueous Metabolite Profiles. Int. J. Mol. Sci. 2024, 25, 2407. https://doi.org/10.3390/ijms25042407
Fuchs AL, Costello SM, Schiller SM, Tripet BP, Copié V. Primary Human M2 Macrophage Subtypes Are Distinguishable by Aqueous Metabolite Profiles. International Journal of Molecular Sciences. 2024; 25(4):2407. https://doi.org/10.3390/ijms25042407
Chicago/Turabian StyleFuchs, Amanda L., Stephanann M. Costello, Sage M. Schiller, Brian P. Tripet, and Valérie Copié. 2024. "Primary Human M2 Macrophage Subtypes Are Distinguishable by Aqueous Metabolite Profiles" International Journal of Molecular Sciences 25, no. 4: 2407. https://doi.org/10.3390/ijms25042407