
Protein Translation in the Pathogenesis of Parkinson’s Disease
 , and
, and
Abstract
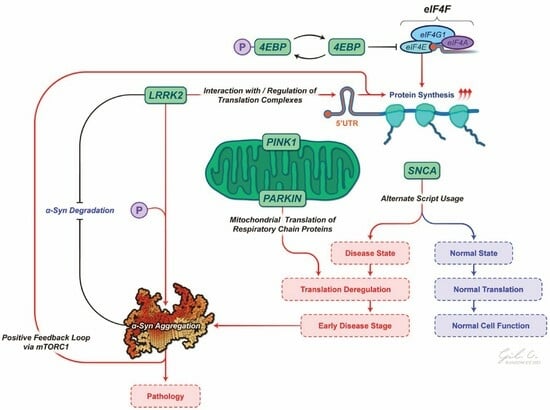
:
1. Introduction
2. How Can Altered Proteostasis Lead to Neuronal Death?
3. Leucine-Rich Repeat Kinase 2 (LRRK2)
3.1. LRRK2 and Its Canonical Roles
3.2. LRRK2 in Parkinson’s Disease and Protein Synthesis
3.3. LRRK2 and Its Interplay with α-Synuclein in Protein Translation
3.4. LRRK2 and Pharmacological Intervention
3.5. Final Thoughts on LRRK2
4. PINK1/PARK2 and Other Molecular Players
4.1. PINK1/Parkin and Their Role in PD Pathogenesis
4.2. Variable Penetrance of eIF4G1 Mutations May Suggest That PD Pathogenesis Only Precipitates in These Cases if Neurons Are Placed under Adequate Stress
4.3. Translation Factors Such as 4E-BPs Regulate the Ability of Cells to Respond to Stress and Proteostasis
5. α-Synuclein: An Active Propagator of PD Pathogenesis
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Van Laar, V.S.; Berman, S.B. Mitochondrial dynamics in Parkinson’s disease. Exp. Neurol. 2009, 218, 247–256. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Niso-Santano, M.; Gonzalez-Polo, R.A.; Fuentes Rodriguez, J.M. Parkinson’s disease: Leucine-rich repeat kinase 2 and autophagy, intimate enemies. Park. Dis. 2012, 2012, 151039. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef]
- Park, Y.; Hoang, Q.Q. Combating Parkinson’s disease-associated toxicity by modulating proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 803–804. [Google Scholar] [CrossRef]
- Kim, J.W.; Yin, X.; Jhaldiyal, A.; Khan, M.R.; Martin, I.; Xie, Z.; Perez-Rosello, T.; Kumar, M.; Abalde-Atristain, L.; Xu, J.; et al. Defects in mRNA Translation in LRRK2-Mutant hiPSC-Derived Dopaminergic Neurons Lead to Dysregulated Calcium Homeostasis. Cell Stem Cell 2020, 27, 633–645. [Google Scholar] [CrossRef]
- Martin, I.; Kim, J.W.; Lee, B.D.; Kang, H.C.; Xu, J.C.; Jia, H.; Stankowski, J.; Kim, M.S.; Zhong, J.; Kumar, M.; et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 2014, 157, 472–485. [Google Scholar] [CrossRef]
- Tain, L.S.; Chowdhury, R.B.; Tao, R.N.; Plun-Favreau, H.; Moisoi, N.; Martins, L.M.; Downward, J.; Whitworth, A.J.; Tapon, N. Drosophila HtrA2 is dispensable for apoptosis but acts downstream of PINK1 independently from Parkin. Cell Death Differ. 2009, 16, 1118–1125. [Google Scholar] [CrossRef]
- Martin, I. Decoding Parkinson’s Disease Pathogenesis: The Role of Deregulated mRNA Translation. J. Park. Dis. 2016, 6, 17–27. [Google Scholar] [CrossRef]
- Kelleher, R.J., III; Bear, M.F. The autistic neuron: Troubled translation? Cell 2008, 135, 401–406. [Google Scholar] [CrossRef]
- Scheper, G.C.; van der Knaap, M.S.; Proud, C.G. Translation matters: Protein synthesis defects in inherited disease. Nat. Rev. Genet. 2007, 8, 711–723. [Google Scholar] [CrossRef]
- Li, W.; Wang, X.; Van Der Knaap, M.S.; Proud, C.G. Mutations linked to leukoencephalopathy with vanishing white matter impair the function of the eukaryotic initiation factor 2B complex in diverse ways. Mol. Cell. Biol. 2004, 24, 3295–3306. [Google Scholar] [CrossRef]
- Satterfield, T.F.; Pallanck, L.J. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum. Mol. Genet. 2006, 15, 2523–2532. [Google Scholar] [CrossRef]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramirez-Jarquin, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021, 12, 1461. [Google Scholar] [CrossRef]
- Glineburg, M.R.; Todd, P.K.; Charlet-Berguerand, N.; Sellier, C. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in Fragile X Tremor Ataxia Syndrome. Brain Res. 2018, 1693, 43–54. [Google Scholar] [CrossRef]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef]
- Galloway, J.N.; Nelson, D.L. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 2009, 4, 785. [Google Scholar] [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef]
- Taymans, J.M.; Nkiliza, A.; Chartier-Harlin, M.C. Deregulation of protein translation control, a potential game-changing hypothesis for Parkinson’s disease pathogenesis. Trends Mol. Med. 2015, 21, 466–472. [Google Scholar] [CrossRef]
- Mamelak, M. Parkinson’s Disease, the Dopaminergic Neuron and Gammahydroxybutyrate. Neurol. Ther. 2018, 7, 5–11. [Google Scholar] [CrossRef]
- Mutez, E.; Nkiliza, A.; Belarbi, K.; de Broucker, A.; Vanbesien-Mailliot, C.; Bleuse, S.; Duflot, A.; Comptdaer, T.; Semaille, P.; Blervaque, R.; et al. Involvement of the immune system, endocytosis and EIF2 signaling in both genetically determined and sporadic forms of Parkinson’s disease. Neurobiol. Dis. 2014, 63, 165–170. [Google Scholar] [CrossRef]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Hershey, J.W. Translational control in mammalian cells. Annu. Rev. Biochem. 1991, 60, 717–755. [Google Scholar] [CrossRef]
- Kthiri, F.; Gautier, V.; Le, H.T.; Prere, M.F.; Fayet, O.; Malki, A.; Landoulsi, A.; Richarme, G. Translational defects in a mutant deficient in YajL, the bacterial homolog of the parkinsonism-associated protein DJ-1. J. Bacteriol. 2010, 192, 6302–6306. [Google Scholar] [CrossRef]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Estrada, A.A.; Sweeney, Z.K. Chemical Biology of Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitors. J. Med. Chem. 2015, 58, 6733–6746. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Kinkl, N.; Schumacher, A.; Braun, R.J.; O’Neill, E.; Meitinger, T.; Kolch, W.; Prokisch, H.; Ueffing, M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006, 15, 223–232. [Google Scholar] [CrossRef]
- Biskup, S.; Moore, D.J.; Celsi, F.; Higashi, S.; West, A.B.; Andrabi, S.A.; Kurkinen, K.; Yu, S.W.; Savitt, J.M.; Waldvogel, H.J.; et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 2006, 60, 557–569. [Google Scholar] [CrossRef]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef]
- Reyniers, L.; Del Giudice, M.G.; Civiero, L.; Belluzzi, E.; Lobbestael, E.; Beilina, A.; Arrigoni, G.; Derua, R.; Waelkens, E.; Li, Y.; et al. Differential protein-protein interactions of LRRK1 and LRRK2 indicate roles in distinct cellular signaling pathways. J. Neurochem. 2014, 131, 239–250. [Google Scholar] [CrossRef]
- Dorval, V.; Hebert, S.S. LRRK2 in Transcription and Translation Regulation: Relevance for Parkinson’s Disease. Front. Neurol. 2012, 3, 12. [Google Scholar] [CrossRef]
- Gehrke, S.; Imai, Y.; Sokol, N.; Lu, B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 2010, 466, 637–641. [Google Scholar] [CrossRef]
- Mills, R.D.; Mulhern, T.D.; Cheng, H.C.; Culvenor, J.G. Analysis of LRRK2 accessory repeat domains: Prediction of repeat length, number and sites of Parkinson’s disease mutations. Biochem. Soc. Trans. 2012, 40, 1086–1089. [Google Scholar] [CrossRef]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef]
- Goldwurm, S.; Zini, M.; Mariani, L.; Tesei, S.; Miceli, R.; Sironi, F.; Clementi, M.; Bonifati, V.; Pezzoli, G. Evaluation of LRRK2 G2019S penetrance: Relevance for genetic counseling in Parkinson disease. Neurology 2007, 68, 1141–1143. [Google Scholar] [CrossRef]
- Bouhnik, J.; Savoie, F.; Michaud, A.; Baussant, T.; Alhenc-Gelas, F.; Corvol, P. Effect of sex hormones on plasma T-kininogen in the rat. Life Sci. 1989, 44, 1859–1866. [Google Scholar] [CrossRef]
- Deshpande, P.; Flinkman, D.; Hong, Y.; Goltseva, E.; Siino, V.; Sun, L.; Peltonen, S.; Elo, L.L.; Kaasinen, V.; James, P.; et al. Protein synthesis is suppressed in sporadic and familial Parkinson’s disease by LRRK2. FASEB J. 2020, 34, 14217–14233. [Google Scholar] [CrossRef]
- Flinkman, D.; Hong, Y.; Gnjatovic, J.; Deshpande, P.; Ortutay, Z.; Peltonen, S.; Kaasinen, V.; James, P.; Coffey, E. Regulators of proteostasis are translationally repressed in fibroblasts from patients with sporadic and LRRK2-G2019S Parkinson’s disease. NPJ Park. Dis. 2023, 9, 20. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Stankowski, J.N.; Lee, B.D.; Grima, J.C.; Chen, G.; Ko, H.S.; Lee, Y.; Swing, D.; et al. Overexpression of Parkinson’s Disease-Associated Mutation LRRK2 G2019S in Mouse Forebrain Induces Behavioral Deficits and alpha-Synuclein Pathology. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Majbour, N.K.; Aasly, J.O.; Hustad, E.; Thomas, M.A.; Vaikath, N.N.; Elkum, N.; van de Berg, W.D.J.; Tokuda, T.; Mollenhauer, B.; Berendse, H.W.; et al. CSF total and oligomeric alpha-Synuclein along with TNF-alpha as risk biomarkers for Parkinson’s disease: A study in LRRK2 mutation carriers. Transl. Neurodegener. 2020, 9, 15. [Google Scholar] [CrossRef]
- Zhao, Y.; Keshiya, S.; Perera, G.; Schramko, L.; Halliday, G.M.; Dzamko, N. LRRK2 kinase inhibitors reduce alpha-synuclein in human neuronal cell lines with the G2019S mutation. Neurobiol. Dis. 2020, 144, 105049. [Google Scholar] [CrossRef]
- Koukouraki, P.; Doxakis, E. Constitutive translation of human alpha-synuclein is mediated by the 5′-untranslated region. Open Biol. 2016, 6, 160022. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.R.; Castro, S.; Drolet, R.; Hatcher, N.G.; Yao, L.; Smith, S.M.; Keeney, M.T.; Di Maio, R.; Kofler, J.; et al. LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104626. [Google Scholar] [CrossRef]
- Kavanagh, M.E.; Doddareddy, M.R.; Kassiou, M. The development of CNS-active LRRK2 inhibitors using property-directed optimisation. Bioorg. Med. Chem. Lett. 2013, 23, 3690–3696. [Google Scholar] [CrossRef]
- Baptista, M.A.S.; Merchant, K.; Barrett, T.; Bhargava, S.; Bryce, D.K.; Ellis, J.M.; Estrada, A.A.; Fell, M.J.; Fiske, B.K.; Fuji, R.N.; et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 2020, 12, eaav0820. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Vissers, M.; Daryani, V.M.; Diaz, D.; Goo, M.S.; Chen, J.J.; Maciuca, R.; Fraser, K.; Mabrouk, O.S.; et al. LRRK2 Inhibition by BIIB122 in Healthy Participants and Patients with Parkinson’s Disease. Mov. Disord. 2023, 38, 386–398. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, H.; Raman, I.; Yan, M.; Chen, Q.; Li, Q.Z. Peripheral Blood Mononuclear Cell Gene Expression in Chronic Obstructive Pulmonary Disease: miRNA and mRNA Regulation. J. Inflamm. Res. 2022, 15, 2167–2180. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef]
- Celardo, I.; Lehmann, S.; Costa, A.C.; Loh, S.H.; Miguel Martins, L. dATF4 regulation of mitochondrial folate-mediated one-carbon metabolism is neuroprotective. Cell Death Differ. 2017, 24, 638–648. [Google Scholar] [CrossRef]
- Bond, S.; Lopez-Lloreda, C.; Gannon, P.J.; Akay-Espinoza, C.; Jordan-Sciutto, K.L. The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2alpha in Neurodegeneration. J. Neuropathol. Exp. Neurol. 2020, 79, 123–143. [Google Scholar] [CrossRef]
- Nies, Y.H.; Mohamad Najib, N.H.; Lim, W.L.; Kamaruzzaman, M.A.; Yahaya, M.F.; Teoh, S.L. MicroRNA Dysregulation in Parkinson’s Disease: A Narrative Review. Front. Neurosci. 2021, 15, 660379. [Google Scholar] [CrossRef]
- Wang, R.; Yao, J.; Gong, F.; Chen, S.; He, Y.; Hu, C.; Li, C. miR-29c-3p regulates TET2 expression and inhibits autophagy process in Parkinson’s disease models. Genes. Cells 2021, 26, 684–697. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, Y.; Li, Z.; Zhu, M.; Wang, Z.; Li, Y.; Xu, T.; Feng, D.; Zhang, S.; Tang, F.; et al. miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol. Res. 2020, 160, 105197. [Google Scholar] [CrossRef]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.X.; Kim, C.; Nelson, P.T.; Springer, W.; Kim, J. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef]
- Koentjoro, B.; Park, J.S.; Ha, A.D.; Sue, C.M. Phenotypic variability of parkin mutations in single kindred. Mov. Disord. 2012, 27, 1299–1303. [Google Scholar] [CrossRef]
- Koentjoro, B.; Park, J.S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef]
- Li, W.; Chen, H.; Li, S.; Lin, G.; Feng, D. Exploring MicroRNAs on NIX-Dependent Mitophagy. Methods Mol. Biol. 2018, 1759, 111–121. [Google Scholar] [CrossRef]
- Sonenberg, N.; Dever, T.E. Eukaryotic translation initiation factors and regulators. Curr. Opin. Struct. Biol. 2003, 13, 56–63. [Google Scholar] [CrossRef]
- Deng, H.; Wu, Y.; Jankovic, J. The EIF4G1 gene and Parkinson’s disease. Acta Neurol. Scand. 2015, 132, 73–78. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Dachsel, J.C.; Vilarino-Guell, C.; Lincoln, S.J.; Lepretre, F.; Hulihan, M.M.; Kachergus, J.; Milnerwood, A.J.; Tapia, L.; Song, M.S.; et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011, 89, 398–406. [Google Scholar] [CrossRef]
- Schulte, E.C.; Mollenhauer, B.; Zimprich, A.; Bereznai, B.; Lichtner, P.; Haubenberger, D.; Pirker, W.; Brucke, T.; Molnar, M.J.; Peters, A.; et al. Variants in eukaryotic translation initiation factor 4G1 in sporadic Parkinson’s disease. Neurogenetics 2012, 13, 281–285. [Google Scholar] [CrossRef]
- Fujioka, S.; Sundal, C.; Strongosky, A.J.; Castanedes, M.C.; Rademakers, R.; Ross, O.A.; Vilarino-Guell, C.; Farrer, M.J.; Wszolek, Z.K.; Dickson, D.W. Sequence variants in eukaryotic translation initiation factor 4-gamma (eIF4G1) are associated with Lewy body dementia. Acta Neuropathol. 2013, 125, 425–438. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Villa, N.; Do, A.; Hershey, J.W.; Fraser, C.S. Human eukaryotic initiation factor 4G (eIF4G) protein binds to eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J. Biol. Chem. 2013, 288, 32932–32940. [Google Scholar] [CrossRef]
- Foeger, N.; Kuehnel, E.; Cencic, R.; Skern, T. The binding of foot-and-mouth disease virus leader proteinase to eIF4GI involves conserved ionic interactions. FEBS J. 2005, 272, 2602–2611. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef]
- Huttenlocher, J.; Kruger, R.; Capetian, P.; Lohmann, K.; Brockmann, K.; Csoti, I.; Klein, C.; Berg, D.; Gasser, T.; Bonin, M.; et al. EIF4G1 is neither a strong nor a common risk factor for Parkinson’s disease: Evidence from large European cohorts. J. Med. Genet. 2015, 52, 37–41. [Google Scholar] [CrossRef]
- Badura, M.; Braunstein, S.; Zavadil, J.; Schneider, R.J. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 18767–18772. [Google Scholar] [CrossRef]
- Ryu, I.; Park, J.H.; An, S.; Kwon, O.S.; Jang, S.K. eIF4GI facilitates the MicroRNA-mediated gene silencing. PLoS ONE 2013, 8, e55725. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Hallegger, M.; Rao, M.S.; Wood, M.J. RISC in PD: The impact of microRNAs in Parkinson’s disease cellular and molecular pathogenesis. Front. Mol. Neurosci. 2013, 6, 40. [Google Scholar] [CrossRef]
- Lan, A.P.; Chen, J.; Zhao, Y.; Chai, Z.; Hu, Y. mTOR Signaling in Parkinson’s Disease. Neuromol. Med. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donze, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [CrossRef]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, B.; Zhang, Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016, 15, 781–786. [Google Scholar] [CrossRef]
- Creus-Muncunill, J.; Badillos-Rodriguez, R.; Garcia-Forn, M.; Masana, M.; Garcia-Diaz Barriga, G.; Guisado-Corcoll, A.; Alberch, J.; Malagelada, C.; Delgado-Garcia, J.M.; Gruart, A.; et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 2019, 142, 3158–3175. [Google Scholar] [CrossRef]
- King, M.A.; Hands, S.; Hafiz, F.; Mizushima, N.; Tolkovsky, A.M.; Wyttenbach, A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol. Pharmacol. 2008, 73, 1052–1063. [Google Scholar] [CrossRef]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J. Park. Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef]
- Gomez-Benito, M.; Granado, N.; Garcia-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wullner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef]
- Grosso Jasutkar, H.; Oh, S.E.; Mouradian, M.M. Therapeutics in the Pipeline Targeting alpha-Synuclein for Parkinson’s Disease. Pharmacol. Rev. 2022, 74, 207–237. [Google Scholar] [CrossRef]
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037. [Google Scholar] [CrossRef]
- Riley, B.E.; Gardai, S.J.; Emig-Agius, D.; Bessarabova, M.; Ivliev, A.E.; Schule, B.; Alexander, J.; Wallace, W.; Halliday, G.M.; Langston, J.W.; et al. Systems-based analyses of brain regions functionally impacted in Parkinson’s disease reveals underlying causal mechanisms. PLoS ONE 2014, 9, e102909. [Google Scholar] [CrossRef]
- Rhinn, H.; Qiang, L.; Yamashita, T.; Rhee, D.; Zolin, A.; Vanti, W.; Abeliovich, A. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 2012, 3, 1084. [Google Scholar] [CrossRef]
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef]
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’Neill, M.J.; Wong, L.F.; et al. Loss of MicroRNA-7 Regulation Leads to alpha-Synuclein Accumulation and Dopaminergic Neuronal Loss In Vivo. Mol. Ther. 2017, 25, 2404–2414. [Google Scholar] [CrossRef]
- Stolzenberg, E.; Berry, D.; Yang, D.; Lee, E.Y.; Kroemer, A.; Kaufman, S.; Wong, G.C.L.; Oppenheim, J.J.; Sen, S.; Fishbein, T.; et al. A Role for Neuronal Alpha-Synuclein in Gastrointestinal Immunity. J. Innate Immun. 2017, 9, 456–463. [Google Scholar] [CrossRef]
- Marreiros, R.; Muller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hansch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Taymans, J.M.; Cookson, M.R. Mechanisms in dominant parkinsonism: The toxic triangle of LRRK2, alpha-synuclein, and tau. Bioessays 2010, 32, 227–235. [Google Scholar] [CrossRef]
- Khan, M.R.; Yin, X.; Kang, S.U.; Mitra, J.; Wang, H.; Ryu, T.; Brahmachari, S.; Karuppagounder, S.S.; Kimura, Y.; Jhaldiyal, A.; et al. Enhanced mTORC1 signaling and protein synthesis in pathologic alpha-synuclein cellular and animal models of Parkinson’s disease. Sci. Transl. Med. 2023, 15, eadd0499. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Yi, S.; Sahni, N.; Loh, K.H.; Auluck, P.K.; Baru, V.; Udeshi, N.D.; Freyzon, Y.; Carr, S.A.; et al. In Situ Peroxidase Labeling and Mass-Spectrometry Connects Alpha-Synuclein Directly to Endocytic Trafficking and mRNA Metabolism in Neurons. Cell Syst. 2017, 4, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Esparcia, P.; Hernandez-Ortega, K.; Koneti, A.; Gil, L.; Delgado-Morales, R.; Castano, E.; Carmona, M.; Ferrer, I. Altered machinery of protein synthesis is region- and stage-dependent and is associated with alpha-synuclein oligomers in Parkinson’s disease. Acta Neuropathol. Commun. 2015, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Neelagandan, N.; Gonnella, G.; Dang, S.; Janiesch, P.C.; Miller, K.K.; Kuchler, K.; Marques, R.F.; Indenbirken, D.; Alawi, M.; Grundhoff, A.; et al. TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 2019, 47, 341–361. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Encoded | Protein Function | Mendelian Inheritance | Proposed Link to Protein Translation in PD |
|---|---|---|---|---|
| EIF4EBP1 | Eukaryotic Translation Initiation Factor 4E-Binding Protein 1 | Regulates EIF4E function, allowing cells to respond rapidly to stressors through altered translation | Inactivation/loss of function results in aberrant de novo protein synthesis and activation of its Drosophila homolog rescues PINK1/Parkin PD phenotypes | |
| EIF4G1 | Eukaryotic Translation Initiation Factor 4 Gamma 1 | Facilitates recognition of the mRNA cap structure and recruitment of mRNA to the ribosome | Late-Onset, Autosomal Dominant PD | Impairs cap-dependent cellular protein synthesis, impairing the ability of cells to respond to stress |
| LRRK2 | Leucine-Rich Repeat Kinase 2 | Large protein kinase is involved in a number of processes, including vesicle trafficking, cytoskeletal dynamics, autophagy, and protein translation | Late-Onset, Autosomal Dominant PD | Increase in global protein synthesis with a disproportionate effect on α-synuclein, as well as phosphorylating α-synuclein, leading to its accumulation and aggregation |
| PARK2 | PARKIN | E3 ubiquitin ligase | Early-Onset, Autosomal Recessive PD | Dysregulates translation of respiratory chain proteins at the mitochondrial outer membrane |
| PINK1 | Pten-Induced Kinase 1 | Mitochondrially targeted kinase, important in mitochondrial quality control | Early-Onset, Autosomal Recessive PD | Dysregulates translation of respiratory chain proteins at the mitochondrial outer membrane |
| SNCA | α-Synuclein | Regulates synaptic vesicle trafficking | Autosomal Dominant PD | Alternate transcript utilized in PD pathogenesis and propagated increased rates of protein translation through interaction with downstream molecular players |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, D.; Khan, M.R.; Dawson, T.M.; Dawson, V.L. Protein Translation in the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 2393. https://doi.org/10.3390/ijms25042393
Ashraf D, Khan MR, Dawson TM, Dawson VL. Protein Translation in the Pathogenesis of Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(4):2393. https://doi.org/10.3390/ijms25042393
Chicago/Turabian StyleAshraf, Daniyal, Mohammed Repon Khan, Ted M. Dawson, and Valina L. Dawson. 2024. "Protein Translation in the Pathogenesis of Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 4: 2393. https://doi.org/10.3390/ijms25042393