
Hub Genes and Pathways Related to Lemon (Citrus limon) Leaf Response to Plenodomus tracheiphilus Infection and Influenced by Pseudomonas mediterranea Biocontrol Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Fungus DNA and Disease Index as Affected by P. mediterranea Treatment
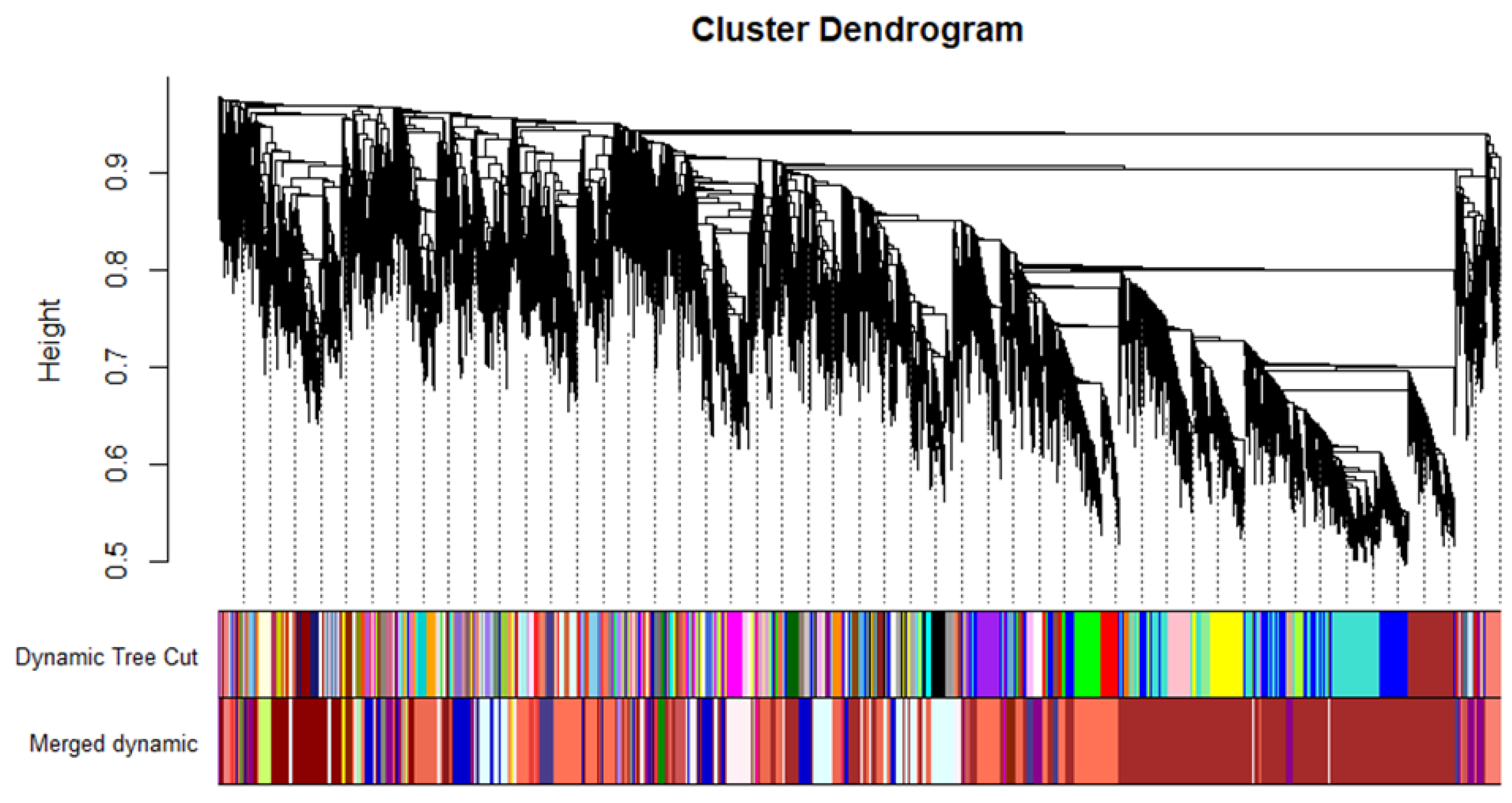
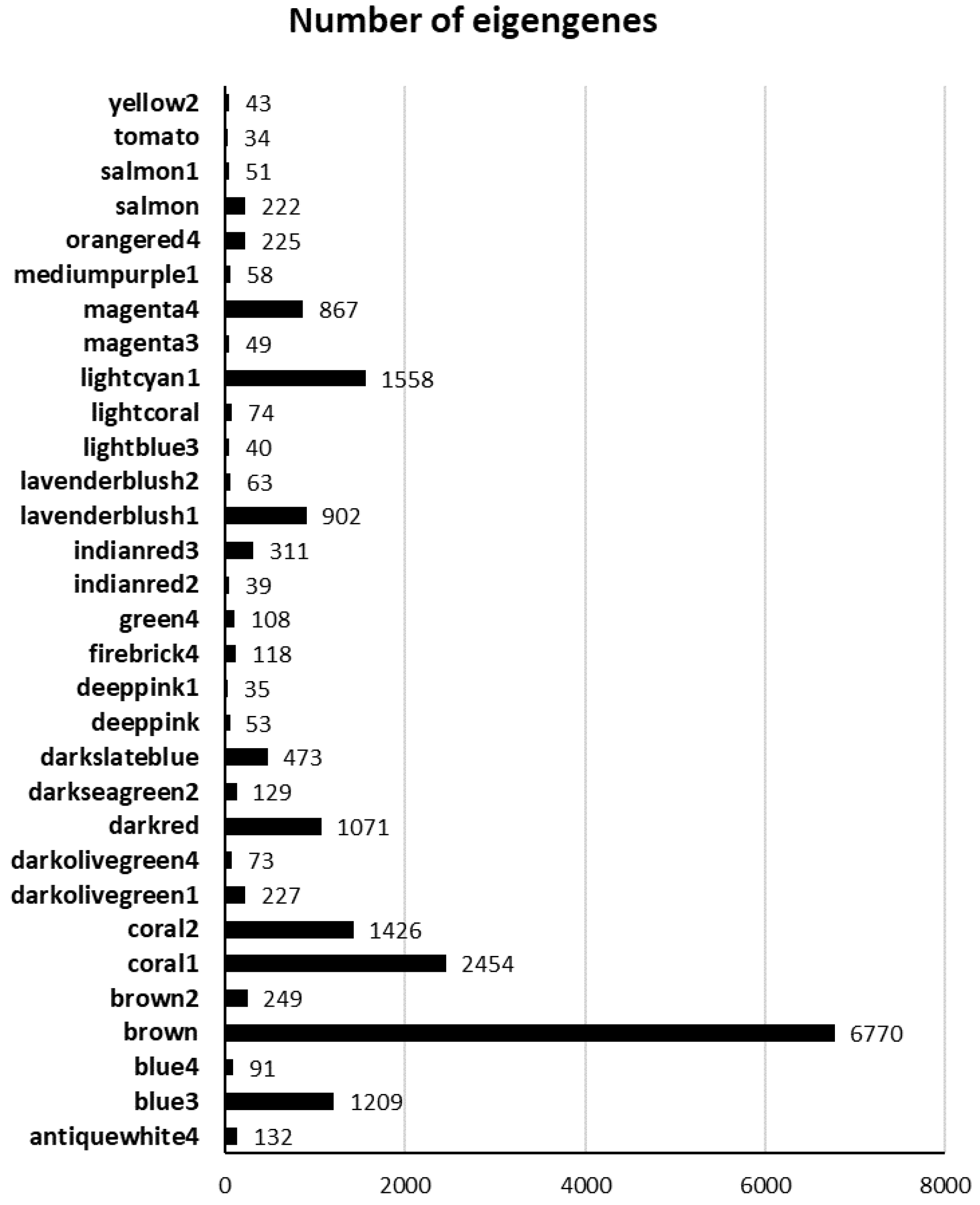
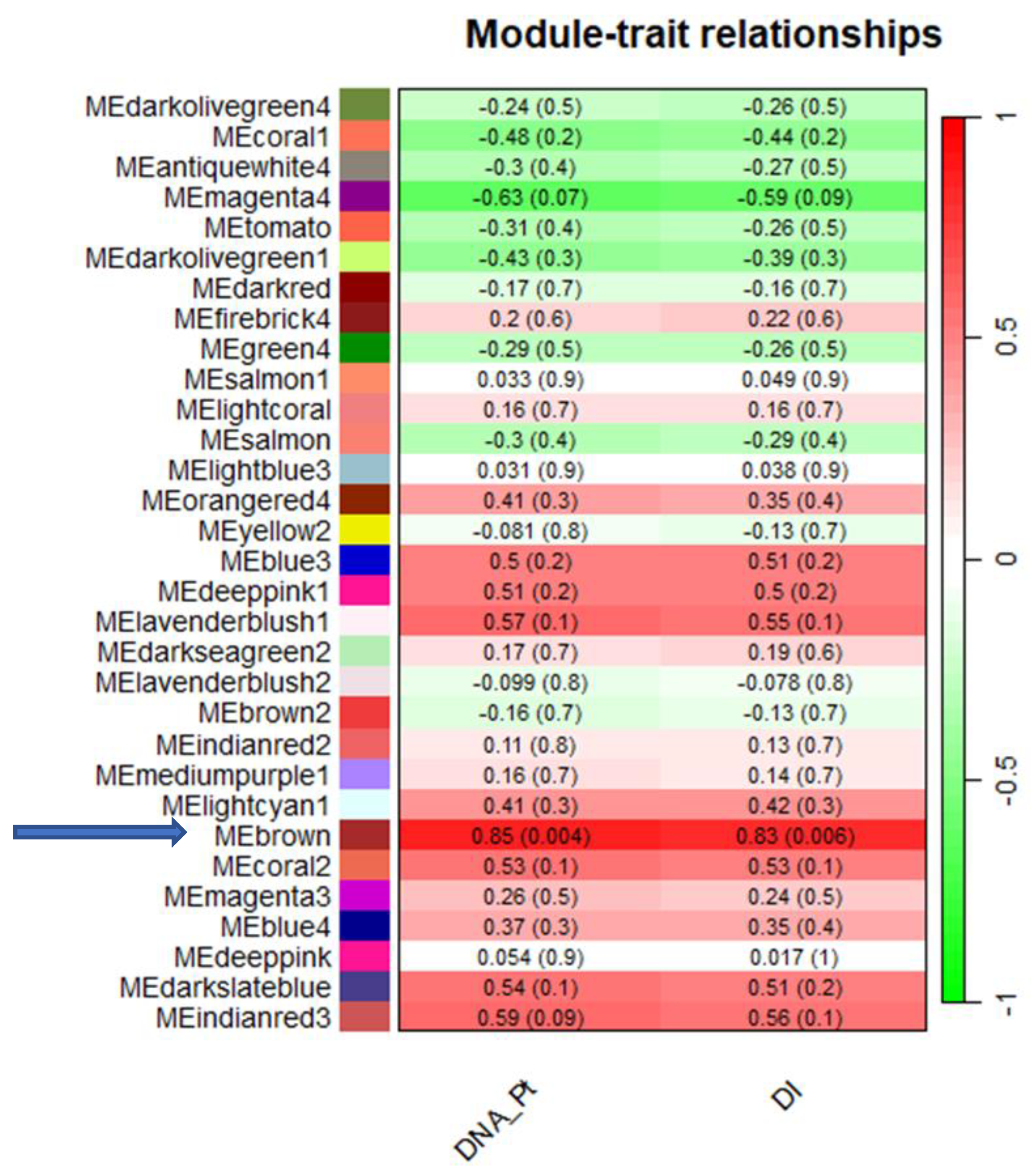
2.2. WCGNA Reveals Gene Modules Associated with P. tracheiphilus DNA and Mal Secco Disease Index
2.3. Functional Enrichment Analysis of the Eigengenes in Brown Module
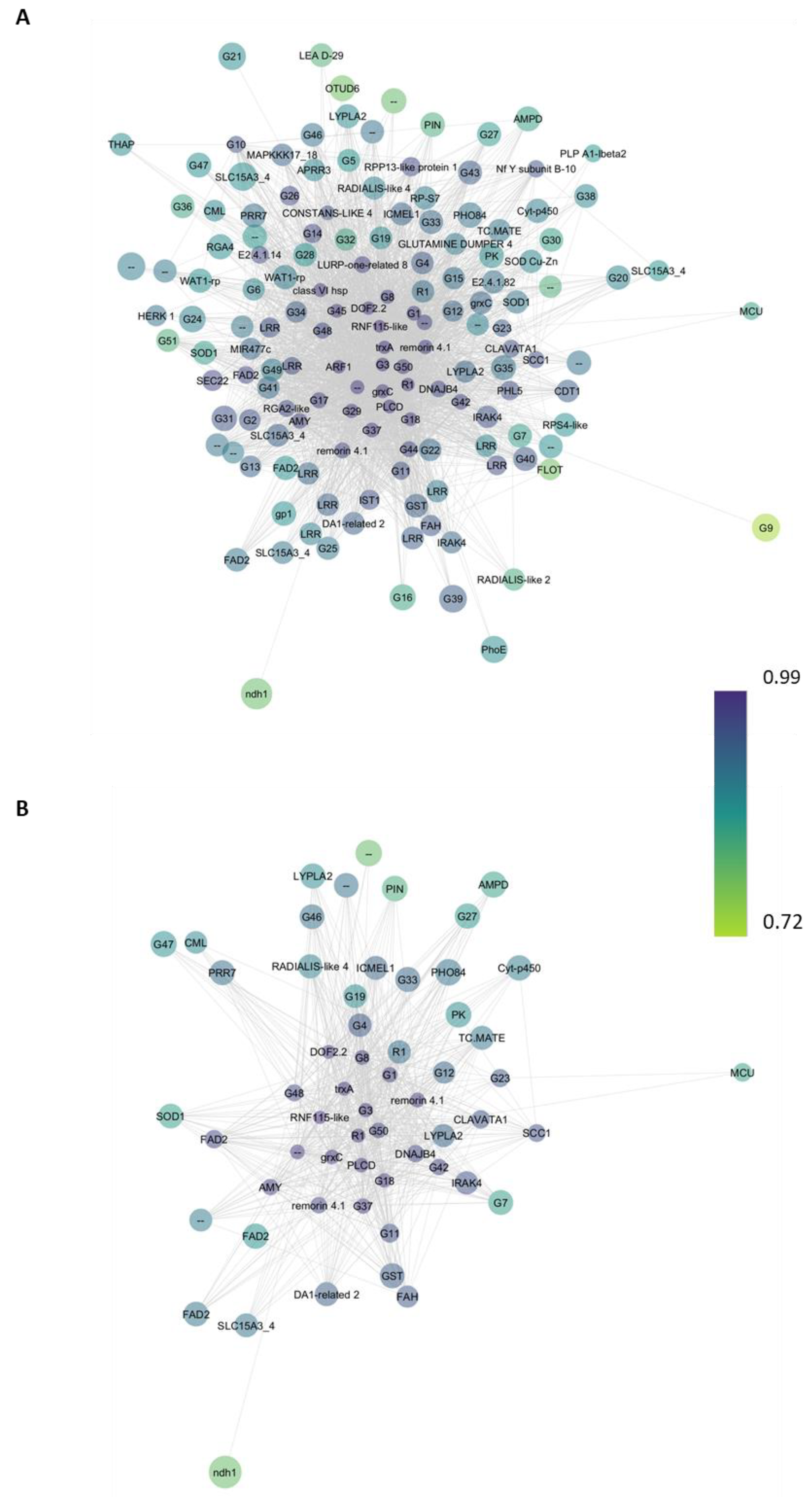
2.4. Functional Analysis of Co-Expression Networks
2.5. Real Time Validation of Hub Genes in Co-Expression Network
3. Discussion
4. Materials and Methods
4.1. Preparation of Pseudomonas mediterranea Cell-Based Bioformulation and of Fungal Pathogen Inoculum
4.2. Transcriptome and Sequencing
4.3. Weighted Correlation Networks Analysis
4.4. Functional Enrichment of Module Genes
4.5. Network Construction
4.6. Validation of Hub Genes by Quantitative Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nigro, F.; Ippolito, A.; Salerno, M.G. Mal secco disease of citrus: A journey through a century of research. J. Plant Pathol. 2011, 93, 523. [Google Scholar]
- Migheli, Q.; Cacciola, S.O.; Balmas, V.; Pane, A.; Ezra, D.; Di San Lio, G.M. Mal secco disease caused by Phoma tracheiphila: A potential threat to lemon production worldwide. Plant Dis. 2009, 93, 852–867. [Google Scholar] [CrossRef]
- Catalano, C.; Di Guardo, M.; Distefano, G.; Caruso, M.; Nicolosi, E.; Deng, Z.; Gentile, A.; La Malfa, S.G. Biotechnological Approaches for Genetic Improvement of Lemon (Citrus limon (L.) Burm. f.) against Mal Secco Disease. Plants 2021, 10, 1002. [Google Scholar] [CrossRef] [PubMed]
- Strano, C.P.; Bella, P.; Licciardello, G.; Caruso, A.; Catara, V. Role of secondary metabolites in the biocontrol activity of Pseudomonas corrugata and Pseudomonas mediterranea. Eur. J. Plant Pathol. 2017, 149, 103. [Google Scholar] [CrossRef]
- Russo, R.; Sicilia, A.; Caruso, M.; Arlotta, C.; Di Silvestro, S.; Gmitter, F.G., Jr.; Nicolosi, E.; Lo Piero, A.R. De Novo transcriptome sequencing of rough lemon leaves (Citrus jambhiri Lush.) in response to Plenodomus tracheiphilus infection. Int. J. Mol. Sci. 2021, 22, 882. [Google Scholar] [CrossRef] [PubMed]
- Di Guardo, M.; Moretto, M.; Moser, M.; Bianco, L.; Gentile, A. De novo assembly of Citrus limon and target-sequence genotyping toward the detection of genes involved in tolerance to ‘mal secco’ disease. Acta Hortic. 2023, 1362, 231–237. [Google Scholar] [CrossRef]
- Catalano, C.; Di Guardo, M.; Distefano, G.; Gentile, A.; La Malfa, S. Genetic Improvement of Citrus Limon (L. Burm f.) for Resistance to Mal Secco Disease. In Genomic Designing for Biotic Stress Resistant Fruit Crops; Kole, C., Ed.; Springer: Cham, Switzerland, 2022; pp. 75–86. [Google Scholar]
- Nigro, F.; Ippolito, A.; Salerno, M.G. Searching for Citrus rootstocks resistant to mal secco disease: A review. Acta Hortic. 2015, 1065, 987–991. [Google Scholar] [CrossRef]
- EPPO Global Database. Available online: https://gd.eppo.int/taxon/DEUTTR (accessed on 17 January 2024).
- Krasnov, H.; Ezra, D.; Bahri, B.A.; Cacciola, S.O.; Meparishvili, G.; Migheli, Q.; Blanket, L. Potential distribution of the citrus Mal Secco disease in the Mediterranean basin under current and future climate conditions. Plant Pathol. 2022, 72, 765–773. [Google Scholar] [CrossRef]
- Conrath, U.; Beckers, G.J.M.; Langenbach, C.J.G.; Jaskiewicz, M.R. Priming for enhanced defense. Annu. Rev. Phytopathol. 2015, 53, 97–119. [Google Scholar] [CrossRef]
- Kalai-Grami, L.; Karkouch, I.; Naili, O.; Slimene, I.B.; Elkahoui, S.; Zekri, R.B.; Touati, I.; Mnari-Hattab, M.; Hajlaoui, M.R.; Limam, F. Production and identification of iturin A lipopeptide from Bacillus methyltrophicus TEB1 for control of Phoma tracheiphila. J. Basic Microbiol. 2016, 56, 864–871. [Google Scholar] [CrossRef]
- Palmieri, D.; Ianiri, G.; Del Grosso, C.; Barone, G.; De Curtis, F.; Castoria, R.; Lima, G. Advances and perspectives in the use of biocontrol agents against fungal plant diseases. Horticulturae 2022, 8, 577. [Google Scholar] [CrossRef]
- Puglisi, I.; Lo Cicero, L.; Lo Piero, A.R. The glutathione S-transferase gene superfamily: An in silico approach to study the post translational regulation. Biodegradation 2013, 24, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Kelbessa, B.G.; Dubey, M.; Catara, V.; Ghadamgahi, F.; Ortiz, R.; Vetukuri, R.R. Potential of plant growth-promoting rhizobacteria to improve crop productivity and adaptation to a changing climate. CABI Rev. 2023, 2023, 1–14. [Google Scholar] [CrossRef]
- Aiello, D.; Leonardi, G.R.; Di Pietro, C.; Vitale, A.; Polizzi, G. A new strategy to improve management of Citrus Mal Secco disease using bioformulates based on Bacillus amyloliquefaciens strains. Plants 2022, 11, 446. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.R.; Polizzi, G.; Vitale, A.; Aiello, D. Efficacy of Biological Control Agents and Resistance Inducer for Control of Mal Secco Disease. Plants 2023, 12, 1735. [Google Scholar] [CrossRef] [PubMed]
- Sicilia, A.; Testa, G.; Santoro, D.F.; Cosentino, S.L.; Lo Piero, A.R. RNASeq analysis of giant cane reveals the leaf transcriptome dynamics under long-term salt stress. BMC Plant Biol. 2019, 19, 355. [Google Scholar] [CrossRef]
- Sicilia, A.; Santoro, D.F.; Testa, G.; Cosentino, S.L.; Lo Piero, A.R. Transcriptional response of giant reed (Arundo donax L.) low ecotype to long-term salt stress by unigene-based RNAseq. Phytochemistry 2020, 177, 112436. [Google Scholar] [CrossRef]
- Kong, P.; Li, X.; Gouker, F.; Hong, C. cDNA Transcriptome of Arabidopsis reveals various defense priming induced by a broad-spectrum biocontrol agent Burkholderia sp. SSG. Int. J. Mol. Sci. 2022, 23, 3151. [Google Scholar] [CrossRef] [PubMed]
- Sicilia, A.; Russo, R.; Caruso, M.; Arlotta, C.; Di Silvestro, S.; Gmitter, F.G., Jr.; Gentile, A.; Nicolosi, E.; Lo Piero, A.R. Transcriptome analysis of Plenodomus tracheiphilus infecting rough lemon (Citrus jambhiri Lush.) indicates a multifaceted strategy during host pathogenesis. Biology 2022, 11, 761. [Google Scholar] [CrossRef]
- Sicilia, A.; Catara, V.; Dimaria, G.; Scialò, E.; Russo, M.; Gentile, A.; Lo Piero, A.R. Transcriptome analysis of lemon leaves (Citrus limon) infected by Plenodomus tracheiphilus reveals the effectiveness of Pseudomonas mediterranea in priming the plant response to mal secco disease. J. Plant Int. 2023, 18, 1–10. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559–566. [Google Scholar] [CrossRef]
- Ju, Z.; Cao, D.; Liang, Y.; Tian, H.; Zhu, B.; Luo, Y. Mining Fruit Ripening-related Transcription Factors in Tomato by Weighted Gene Co-expression Network Analyses (WGCNA). J. Chin. Inst. Food Sci. Technol. 2018, 18, 240–248. [Google Scholar]
- Wu, Q.; Pan, Y.-B.; Su, Y.; Zou, W.; Xu, F.; Sun, T.; Grisham, M.P.; Yang, S.; Xu, L.; Que, Y. Comprehensive and Dynamic Gene Co-Expression Network That Associates with Smut Resistance in Sugarcane. Int. J. Mol. Sci. 2022, 23, 10770. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-X.; Zhang, W.-L.; Jue, D.-W.; Liu, X.; Jiang, Y.-S.; Tang, J.-M. Transcriptome Changes Induced by Botrytis cinerea Stress and Weighted Gene Co-expression Network Analysis (WGCNA) in Actinidia chinensis. Plant Mol. Biol. Rep. 2022, 40, 389–401. [Google Scholar] [CrossRef]
- Azam, M.; Zhang, S.; Li, J.; Ahsan, M.; Agyenim-Boateng, K.G.; Qi, J.; Feng, Y.; Liu, Y.; Li, B.; Qiu, L.; et al. Identification of hub genes regulating isoflavone accumulation in soybean seeds via GWAS and WGCNA approaches. Front. Plant Sci. 2023, 14, 1120498. [Google Scholar] [CrossRef] [PubMed]
- Corke, A.T.K. Biological Control of Plant Pathogens; Baker, K.F., Cook, R.J., Eds.; Freeman: San Francisco, CA, USA, 1974; p. 433. [Google Scholar]
- Raymaekers, K.; Ponet, L.; Holtappels, D.; Berckmans, B.; Cammue, B.P.A. Screening for novel biocontrol agents applicable in plant disease management—A review. Biol. Control 2020, 144, 104240. [Google Scholar] [CrossRef]
- Lahlali, R.; Ezrari, S.; Radouane, N.; Kenfaoui, J.; Esmaeel, Q.; El Hamss, H.; Belabess, Z.; Barka, E.A. Biological control of plant pathogens: A global perspective. Microorganisms 2022, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wu, X.; Tao, Y.; Yan, G.; Wang, X.; Cao, S.; Wang, C.; He, W. Mining Candidate Genes Related to Heavy Metals in Mature Melon (Cucumis melo L.) Peel and Pulp Using WGCNA. Genes 2022, 13, 1767. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, J.; Song, Q.; Cheng, Q.; Tan, Q.; Zhou, Q.; Nong, Z.; Lv, P. Transcriptome and WGCNA reveal hub genes in sugarcane tiller seedlings in response to drought stress. Sci. Rep. 2023, 13, 12823. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, J.; Cao, J.; Ma, X.; Li, W.; Yang, G. Hub Gene Mining and Co-Expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA. Genes 2023, 14, 1598. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Yao, W.; Ge, W.; Jiang, T.; Zhou, B. Transcriptome Sequencing and WGCNA Reveal Key Genes in Response to Leaf Blight in Poplar. Int. J. Mol. Sci. 2023, 24, 10047. [Google Scholar] [CrossRef]
- Iqbal, A.; Khan, R.S. Snakins: Antimicrobial potential and prospects of genetic engineering for enhanced disease resistance in plants. Mol. Biol. Rep. 2023, 50, 8683–8690. [Google Scholar] [CrossRef]
- Nishimura, M.T. Loss of a callose synthase results in salicylic acid—Dependent disease resistance. Science 2003, 301, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Ye, Y.; Hu, C.; Wang, H.; Zheng, L.; Hu, Y.; Sheng, L.; Xing, J.; Jia, W.; Wang, Y.; et al. Exogenous melatonin orchestrates multiple defense responses against Botrytis cinerea in tomato leaves. Plant Stress 2024, 11, 100326. [Google Scholar] [CrossRef]
- Tiwari, S.; Muthusamy, S.K.; Roy, P.; Dalal, M. Genome wide analysis of BREVIS RADIX gene family from wheat (Triticum aestivum): A conserved gene family differentially regulated by hormones and abiotic stresses, Int. J. Biol. Macromol. 2023, 232, 12308. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.A.; Vlot, A.C.; Wildermuth, M.C.; Klessig, D.F. Salicylic acid biosynthesis and metabolism. Arab. Book 2011, 9, e0156. [Google Scholar] [CrossRef]
- Bresson, J.; Doll, J.; Vasseur, F.; Stahl, M.; von Roepenack-Lahaye, E.; Kilian, J.; Stadelhofer, B.; Kremer, J.M.; Kolb, D.; Wenkel, S.; et al. The genetic interaction of REVOLUTA and WRKY53 links plant development, senescence, and immune responses. PLoS ONE 2022, 17, e0254741. [Google Scholar] [CrossRef]
- Jose, J.; Ghantasala, S.; Roy Choudhury, S. Arabidopsis Transmembrane Receptor-Like Kinases (RLKs): A Bridge between Extracellular Signal and Intracellular Regulatory Machinery. Int J Mol Sci. 2020, 3, 4000. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, H.; Zhuang, R.R.; Chen, Y.T.; Deng, Y.; Cai, T.C.; Wang, S.Y.; Liu, Q.Z.; Tang, R.H.; Shan, S.H.; et al. Overexpression of the peanut CLAVATA1-like leucine-rich repeat receptor-like kinase AhRLK1 confers increased resistance to bacterial wilt in tobacco. J. Exp. Bot. 2019, 70, 5407–5421. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Dangl, J. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- van Loon, L.C.; Bakker, P.A.H.M.; Pieterse, C.M.J. Systemic resistance induced by rhizosphere bacteria. Annu. Rev. Phytopathol. 1998, 36, 453–483. [Google Scholar] [CrossRef]
- Fu, Z.Q.; Dong, X. Systemic acquired resistance: Turning local infection into global defense. Annu. Rev. Plant Biol. 2013, 64, 839–863. [Google Scholar] [CrossRef]
- Shine, M.B.; Xiao, X.; Kachroo, P.; Kachroo, A. Signaling mechanisms underlying systemic acquired resistance to microbial pathogens. Plant Sci. 2019, 279, 81–86. [Google Scholar] [CrossRef]
- Choudhary, D.K.; Prakash, A.; Johri, B.N. Induced systemic resistance (ISR) in plants: Mechanism of action. Indian J. Microbiol. 2007, 47, 289–297. [Google Scholar] [CrossRef]
- Dimaria, G.; Mosca, A.; Anzalone, A.; Paradiso, G.; Nicotra, D.; Privitera, G.F.; Pulvirenti, A.; Catara, V. Sour orange microbiome is affected by infections of Plenodomus tracheiphilus causal agent of Citrus mal Secco disease. Agronomy 2023, 13, 654. [Google Scholar] [CrossRef]
- Licciardello, G.; Grasso, F.M.; Bella, P.; Cirvilleri, G.; Grimaldi, V.; Catara, V. Identification and detection of Phoma tracheiphila, causal agent of citrus mal secco disease, by realtime polymerase chain reaction. Plant Dis. 2006, 90, 1523. [Google Scholar] [CrossRef]
- Luisi, N.; De Cicco, V.; Cutuli, G.; Salerno, M. Ricerche su un metodo di studio della patogenicità del mal secco degli agrumi. Ann. Dell’istituto Sper. Per L’agrumicoltura 1979, 9–10, 167–173. [Google Scholar]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar]
- Liu, Y. Bioinformatics: The Impact of Accurate Quantification on Proteomic and Genetic Analysis and Research, 1st ed.; Apple Academic Press: Palm Bay, FL, USA, 2014. [Google Scholar]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sicilia, A.; Russo, R.; Catara, V.; Lo Piero, A.R. Hub Genes and Pathways Related to Lemon (Citrus limon) Leaf Response to Plenodomus tracheiphilus Infection and Influenced by Pseudomonas mediterranea Biocontrol Activity. Int. J. Mol. Sci. 2024, 25, 2391. https://doi.org/10.3390/ijms25042391
Sicilia A, Russo R, Catara V, Lo Piero AR. Hub Genes and Pathways Related to Lemon (Citrus limon) Leaf Response to Plenodomus tracheiphilus Infection and Influenced by Pseudomonas mediterranea Biocontrol Activity. International Journal of Molecular Sciences. 2024; 25(4):2391. https://doi.org/10.3390/ijms25042391
Chicago/Turabian StyleSicilia, Angelo, Riccardo Russo, Vittoria Catara, and Angela Roberta Lo Piero. 2024. "Hub Genes and Pathways Related to Lemon (Citrus limon) Leaf Response to Plenodomus tracheiphilus Infection and Influenced by Pseudomonas mediterranea Biocontrol Activity" International Journal of Molecular Sciences 25, no. 4: 2391. https://doi.org/10.3390/ijms25042391