

Beneficial Impact of Eicosapentaenoic Acid on the Adverse Effects Induced by Palmitate and Hyperglycemia on Healthy Rat Chondrocyte
 , , and
, , and
Abstract
:1. Introduction
2. Results
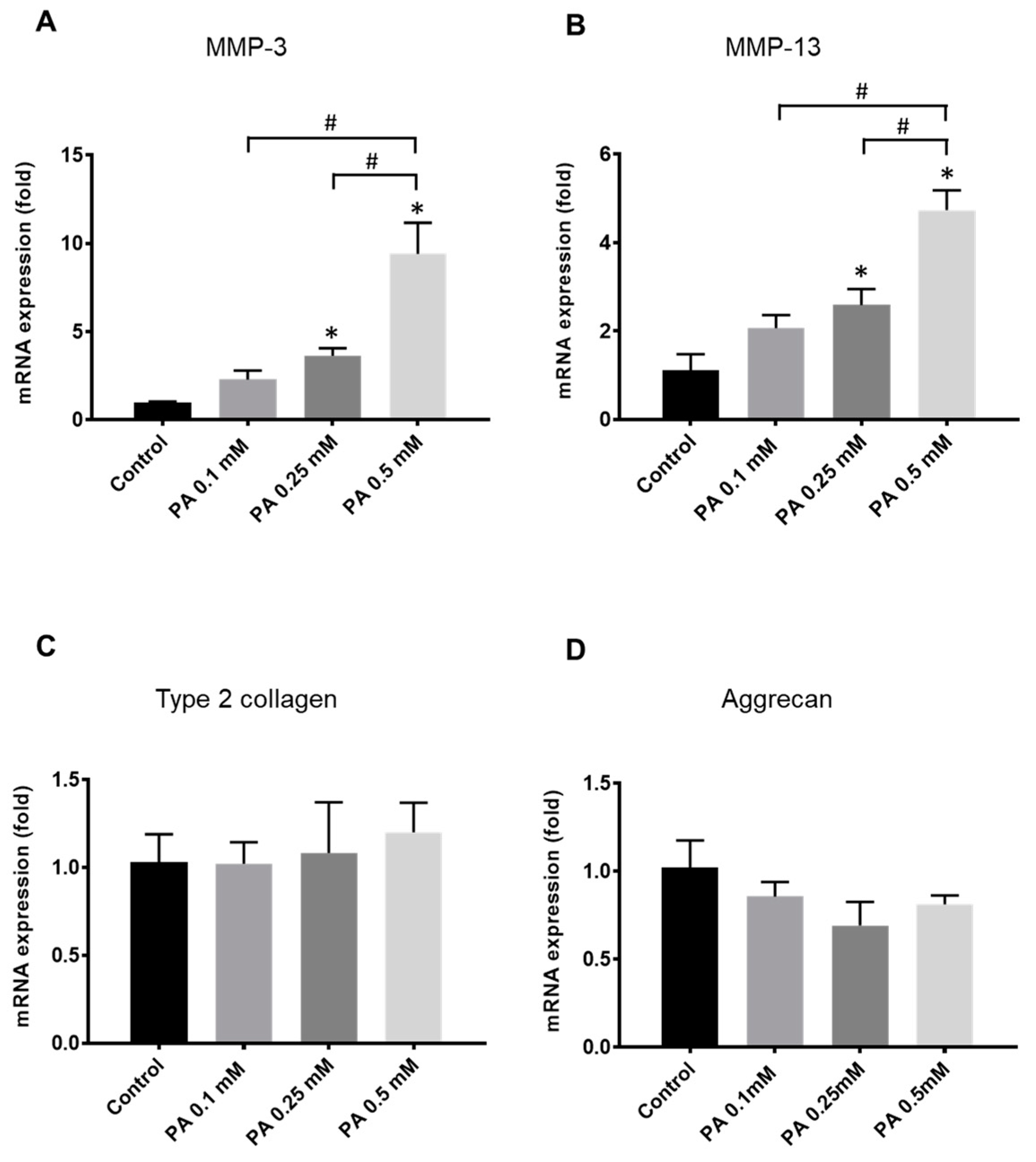
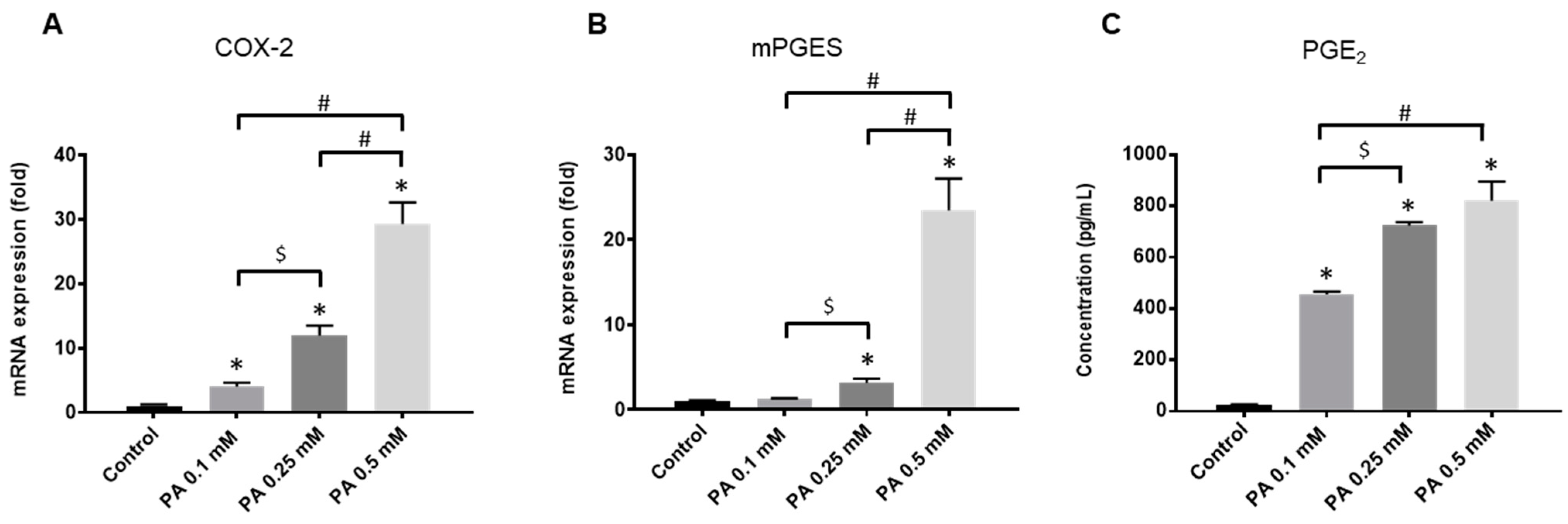
2.1. Rat Chondrocyte Response to PA
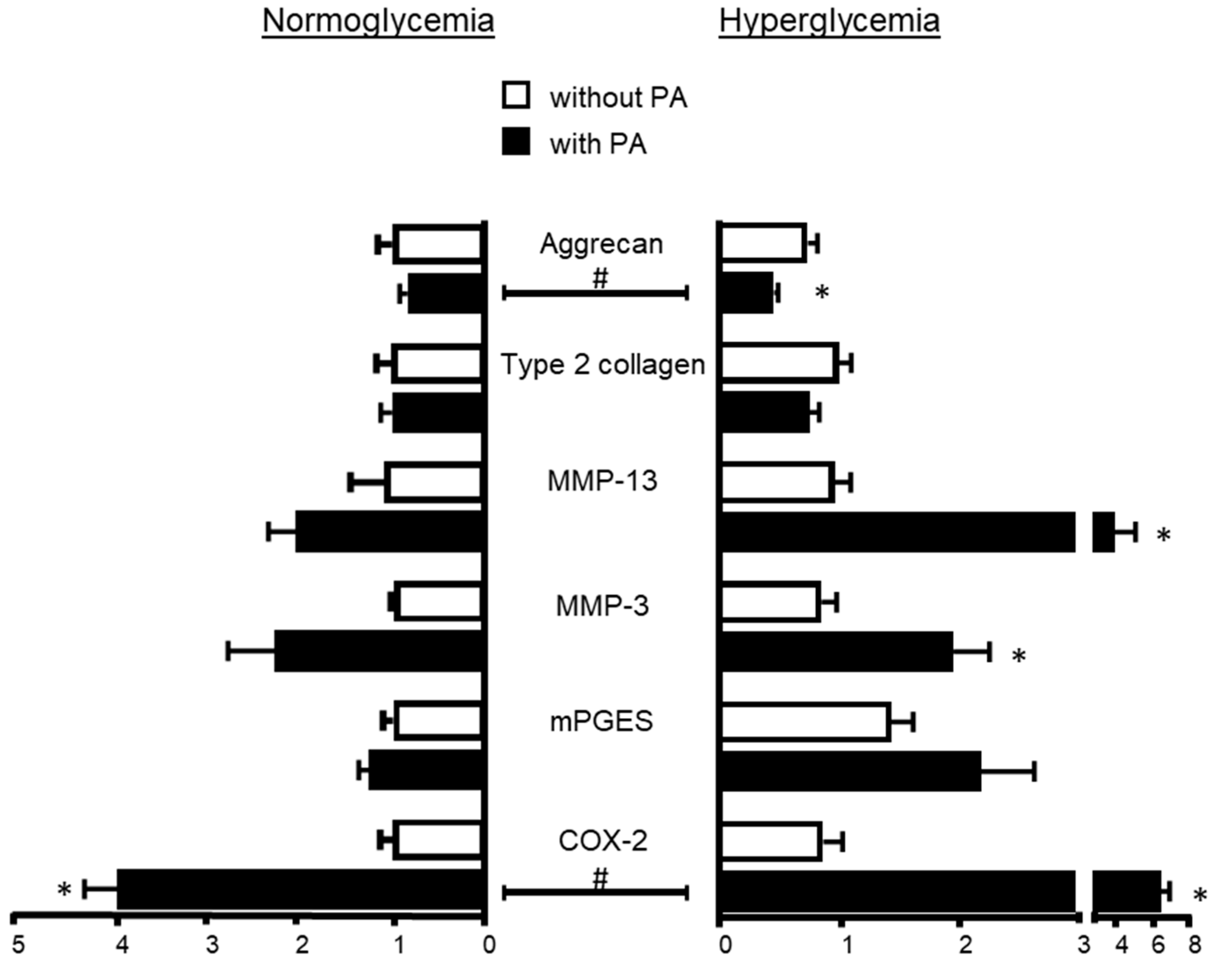
2.2. Influence of Hyperglycemia on PA Effects
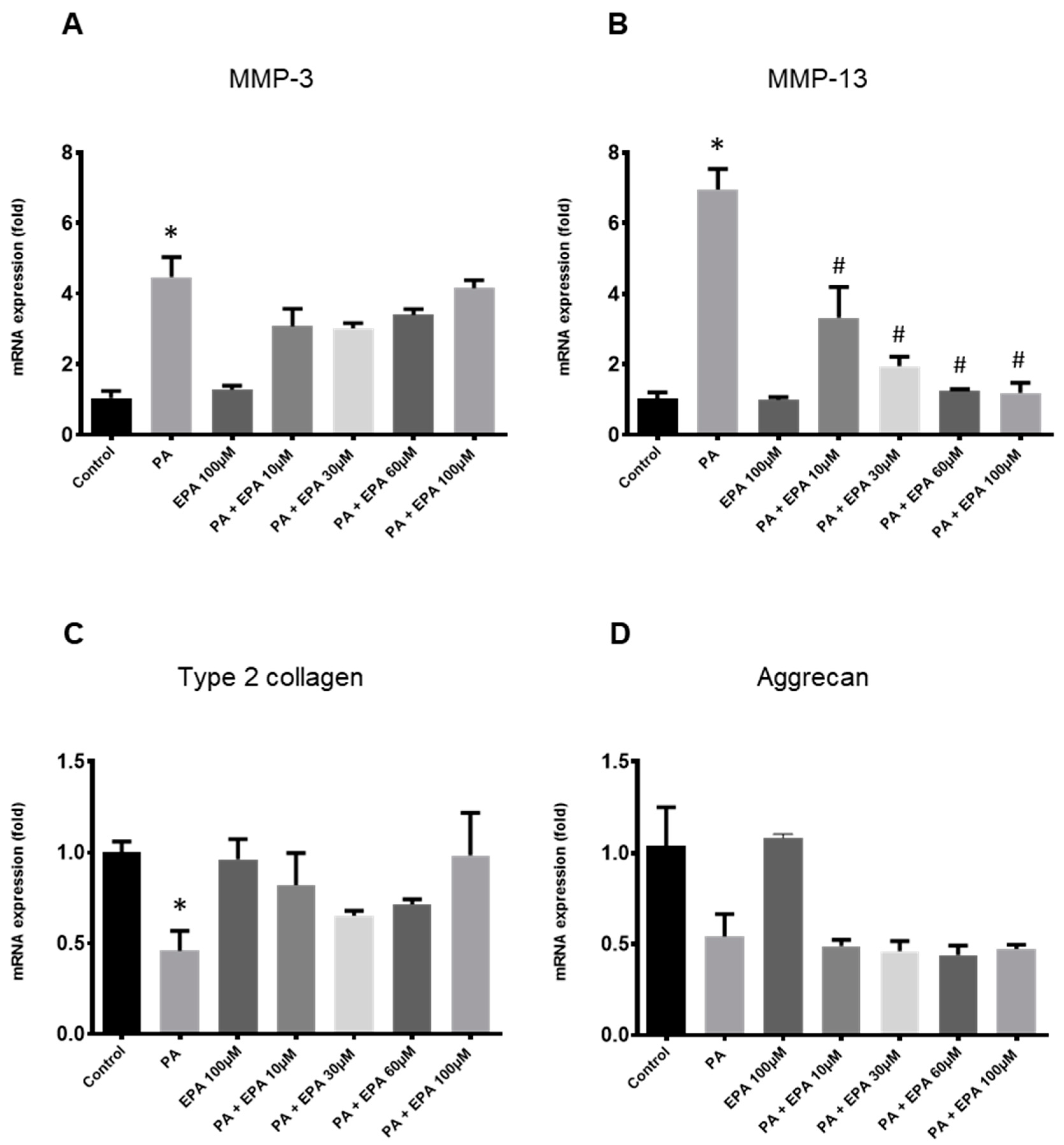
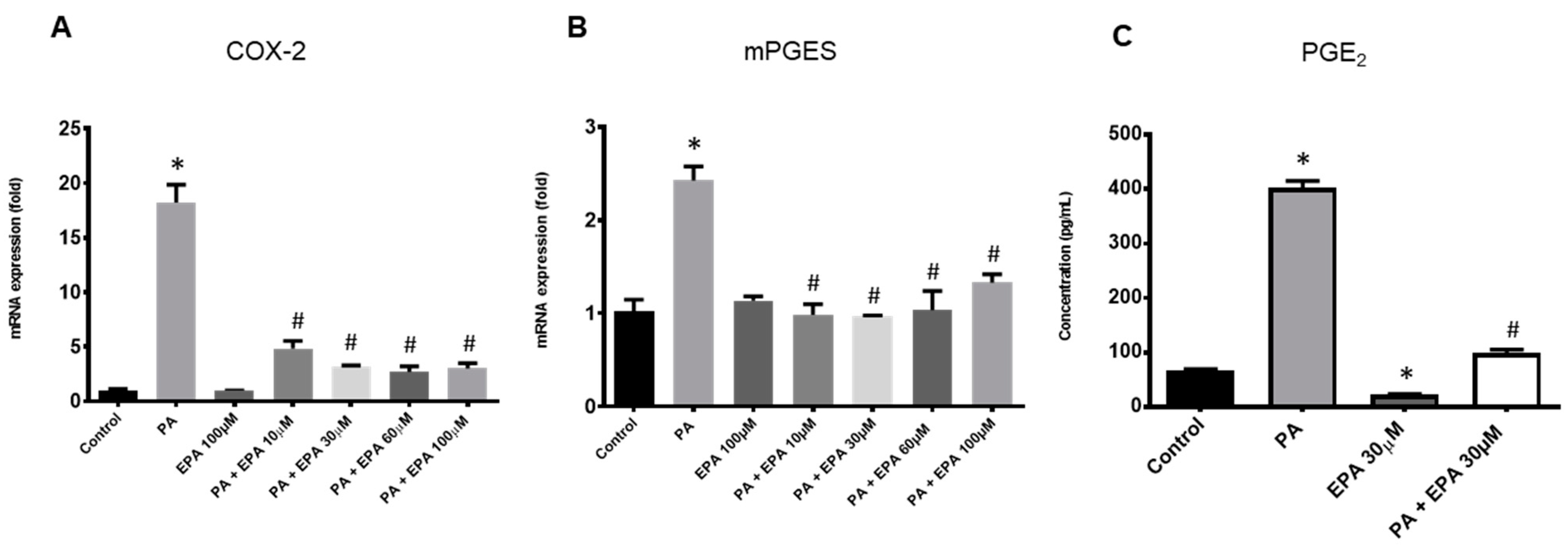
2.3. Effects of EPA on Chondrocyte Response to Both PA and Hyperglycemia
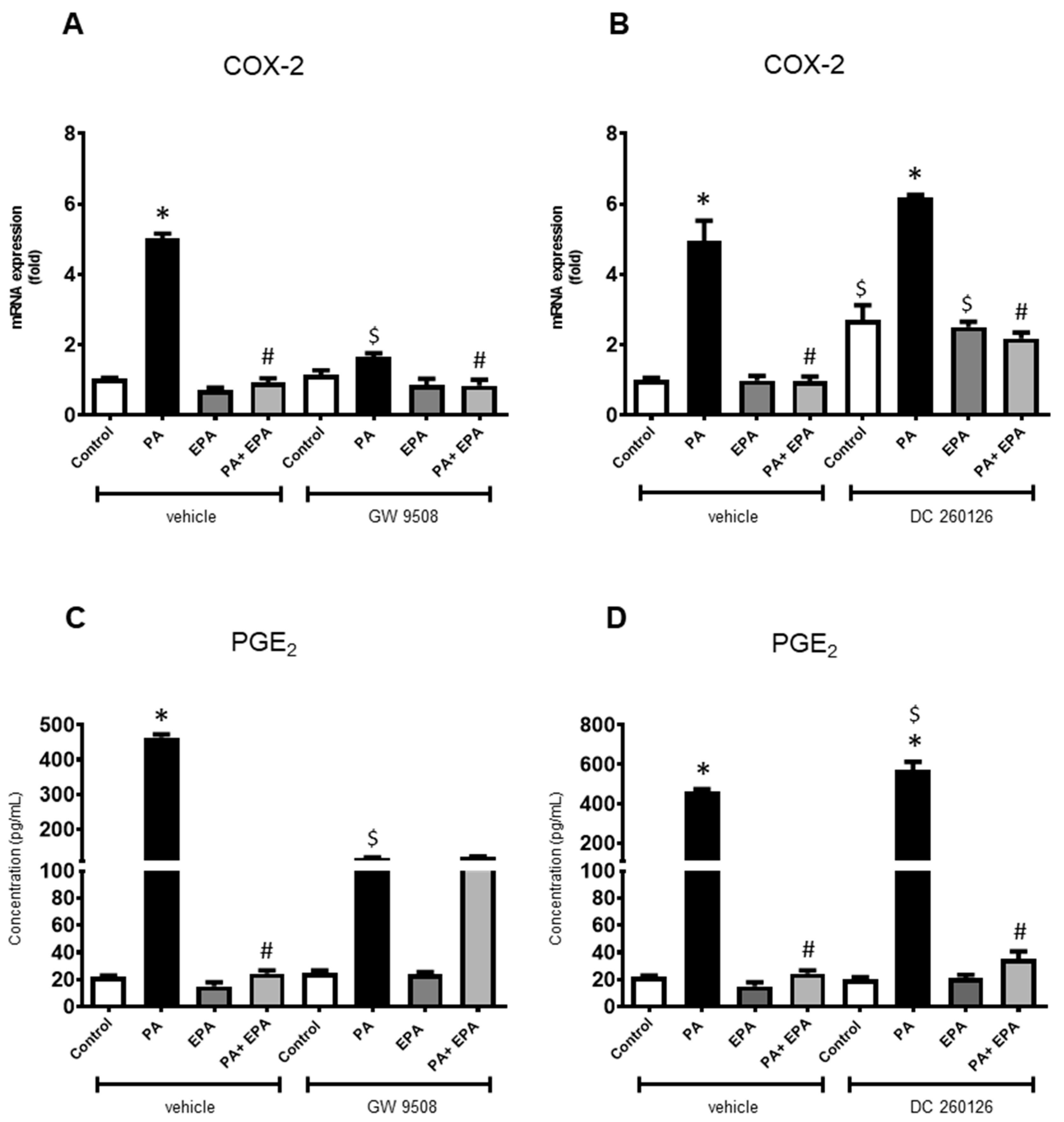
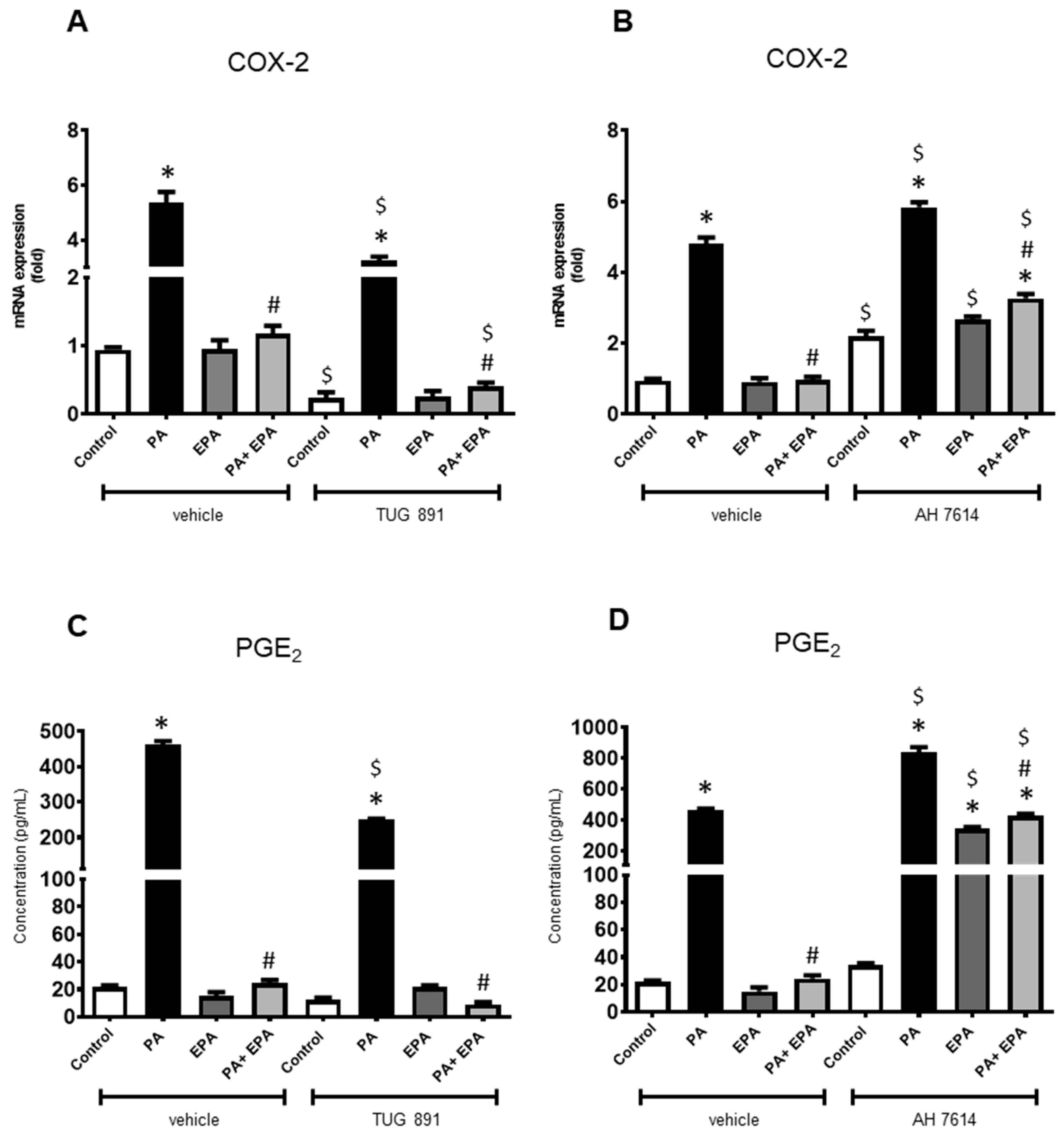
2.4. Identification of Receptors Involved in PA and EPA Effects
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Rat Chondrocytes
4.2. Treatment of Rat Chondrocytes with Fatty Acids, Glucose and FFAR Ligands
4.3. RNA Isolation and Real-Time Polymerase Chain Reaction
4.4. PGE2 Determination by Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Katz, J.N.; Arant, K.R.; Loeser, R.F. Diagnosis and treatment of hip and knee osteoarthritis: A review. JAMA 2021, 325, 568–578. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Puenpatom, R.A.; Victor, T.W. Increased prevalence of metabolic syndrome in individuals with osteoarthritis: An analysis of NHANES III data. Postgrad. Med. 2009, 121, 9–20. [Google Scholar] [CrossRef]
- Yoshimura, N.; Muraki, S.; Oka, H.; Tanaka, S.; Kawaguchi, H.; Nakamura, K.; Akune, T. Accumulation of metabolic risk factors such as overweight, hypertension, dyslipidaemia, and impaired glucose tolerance raises the risk of occurrence and progression of knee osteoarthritis: A 3-year follow-up of the ROAD study. Osteoarthr. Cartil. 2012, 20, 1217–1226. [Google Scholar] [CrossRef]
- Berenbaum, F.; Griffin, T.M.; Liu-Bryan, R. Metabolic regulation of inflammation in osteoarthritis. Arthritis Rheumatol. 2017, 69, 9–21. [Google Scholar] [CrossRef]
- Engstrom, G.; Gerhardsson de Verdier, M.; Rollof, J.; Nilsson, P.M.; Lohmander, L.S. C-reactive protein, metabolic syndrome and incidence of severe hip and knee osteoarthritis: A population-based cohort study. Osteoarthr. Cartil. 2009, 17, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, E.; Nelissen, R.G.; Ioan-Facsinay, A.; Stojanovic-Susulic, V.; DeGroot, J.; van Osch, G.; Middeldorp, S.; Huizinga, T.W.J.; Kloppenburg, M. Association between weight or body mass index and hand osteoarthritis: A systematic review. Ann. Rheum. Dis. 2010, 69, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Visser, A.W.; Ioan-Facsinay, A.; de Mutsert, R.; Widya, R.L.; Loef, M.; de Roos, A.; le Cessie, S.; den Heijer, M.; Rosendaal, F.R.; Kloppenburg, M. NEO Study Group. Adiposity and hand osteoarthritis: The Netherlands Epidemiology of Obesity study. Arthritis Res. Ther. 2014, 16, R19. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, M.A.; Cutolo, M. Plasma glucose concentration in symptomatic osteoarthritis: A clinical and epidemiological survey. Clin. Exp. Rheumatol. 1990, 8, 251–257. [Google Scholar] [PubMed]
- Sturmer, T.; Brenner, H.; Brenner, R.E.; Gunther, K.P. Non-insulin dependent diabetes mellitus (NIDDM) and patterns of osteoarthritis. The Ulm osteoarthritis study. Scand. J. Rheumatol. 2001, 30, 169–171. [Google Scholar] [PubMed]
- Frey, M.I.; Barrett-Connor, E.; Sledge, P.A.; Schneider, D.L.; Weisman, M.H. The effect of noninsulin dependent diabetes mellitus on the prevalence of clinical osteoarthritis. A population based study. J. Rheumatol. 1996, 23, 716–722. [Google Scholar] [PubMed]
- Yammani, R.R.; Carlson, C.S.; Bresnick, A.R.; Loeser, R.F. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.E.; Bruckner, P.; Pujol, J.P. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthr. Cartil. 2003, 11, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Harasymowicz, N.S.; Dicks, A.; Wu, C.L.; Guilak, F. Physiologic and pathologic effects of dietary free fatty acids on cells of the joint. Ann. N. Y. Acad. Sci. 2019, 1440, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Van de Vyver, A.; Clockaerts, S.; van de Lest, C.H.A.; Wei, W.; Verhaar, J.; Van Osch, G.J.V.M.; Bastiaansen-Jenniskens, Y.M. Synovial fluid fatty acid profiles differ between osteoarthritis and healthy patients. Cartilage 2020, 11, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Hwang, J.; Kim, J.; Ahn, J.K.; Cha, H.S.; Kim, K.H. Metabolite profiles of synovial fluid change with the radiographic severity of knee osteoarthritis. Jt. Bone Spine 2017, 84, 605–610. [Google Scholar] [CrossRef]
- Loef, M.; van de Stadt, L.; Böhringer, S.; Bay-Jensen, A.C.; Mobasheri, A.; Larkin, J.; Lafeber, F.P.J.G.; Blanco, F.J.; Haugen, I.K.; Berenbaum, F.; et al. The association of the lipid profile with knee and hand osteoarthritis severity: The IMI-APPROACH cohort. Osteoarthr. Cartil. 2022, 30, 1062–1069. [Google Scholar] [CrossRef]
- Berta Cillero-Pastor, B.; Eijkel, G.; Kiss, A.; Blanco, F.J.; Heeren, R.M.A. Time-of-flight secondary ion mass spectrometry-based molecular distribution distinguishing healthy and osteoarthritic human cartilage. Anal. Chem. 2012, 84, 8909–8916. [Google Scholar] [CrossRef]
- Lippiello, L.; Walsh, T.; Fienhold, M. The association with lipids abnormalities with tissue pathology in human osteoarthritic articular cartilage. Metabolism 1991, 40, 571–576. [Google Scholar] [CrossRef]
- Gkretsi, V.; Simopoulou, T.; Tsezou, A. Lipid metabolism and osteoarthritis: Lessons from atherosclerosis. Prog. Lipid Res. 2011, 50, 133–140. [Google Scholar] [CrossRef]
- Wu, C.L.; Jain, D.; McNeill, J.N.; Little, D.; Anderson, J.A.; Huebner, J.L.; Kraus, W.B.; Rodriguiz, R.M.; Wetsel, W.C.; Guilak, F. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann. Rheum. Dis. 2015, 74, 2076–2083. [Google Scholar] [CrossRef]
- Sekar, S.; Shafie, S.R.; Prasadam, I.; Crawford, R.; Panchal, S.K.; Brown, L.; Xiao, Y. Saturated fatty acids induce development of both metabolic syndrome and osteoarthritis in rats. Sci. Rep. 2017, 7, 46457. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Driban, J.B.; Xu, C.; Lapane, K.L.; McAlindon, T.E.; Eaton, C.B. Dietary fat intake and radiographic progression of knee osteoarthritis: Data from the osteoarthritis initiative. Arthritis Care Res. 2017, 69, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Knott, L.; Avery, N.C.; Hollander, A.P.; Tarlton, J.F. Regulation of osteoarthritis by omega-3 (n-3) polyunsaturated fatty acids in a naturally occurring model of disease. Osteoarthr. Cartil. 2011, 19, 1150–1157. [Google Scholar] [CrossRef]
- Huang, M.; Wang, L.; Zhang, Z.M.; Chen, T.; Jia, C.; Wang, Y.; Zhen, X.; Huang, B.; Yan, B.; Chen, Y.; et al. Enhancement of the synthesis of n-3 PUFAs in fat-1transgenic mice inhibits mTORC1 signaling and delays surgically induced osteoarthritis in comparison with wild-type mice. Ann. Rheum. Dis. 2014, 73, 1719–1727. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free fatty acid receptors in health and disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Kim, I.C.; Cohen, A.S. Synovial fluid fatty acid composition in patients with rheumatoid arthritis, gout and degenerative joint disease. Proc. Soc. Exp. Biol. Med. 1966, 123, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Baudart, P.; Louati, K.; Marcelli, C.; Brenbaum, F.; Sellam, J. Association between osteoarthritis and dyslipidemia: A systemic literature review and meta-analysis. RMD Open 2017, 3, e000442. [Google Scholar] [CrossRef] [PubMed]
- Meessen, J.M.T.A.; Saberi-Hosnijeh, F.; Bomer, N.; den Hollander, W.; van der Bom, J.G.; van Hilten, J.A.; van Spil, W.E.; So-Osman, C.; Uitterlinden, A.G.; Kloppenburg, M.; et al. Serum fatty acid chain length associates with prevalent symptomatic end-stage osteoarthritis, independent of BMI. Sci. Rep. 2020, 10, 15549. [Google Scholar] [CrossRef]
- Louer, C.R.; Furman, B.D.; Huebner, J.L.; Kraus, V.B.; Olson, S.A.; Guilak, F. Diet-induced obesity significantly increases the severity of posttraumatic arthritis in mice. Arthritis Rheum. 2012, 64, 3220–3230. [Google Scholar] [CrossRef]
- Mooney, R.A.; Sampson, E.R.; Lerea, J.; Rosier, R.N.; Zuscik, M.J. High-fat diet accelerates progression of osteoarthritis after meniscal/ligamentous injury. Arthritis Res. Ther. 2011, 13, R198. [Google Scholar] [CrossRef]
- Mustonen, A.M.; Lehmonen, N.; Paakkonen, T.; Raekallio, M.; Käkelä, R.; Niemelä, T.; Mykkänen, A.; Sihvo, S.P.; Nieminen, P. Equine osteoarthritis modifies fatty acid signatures in synovial fluid and its extracellular vesicles. Arthritis Res. Ther. 2023, 25, 39. [Google Scholar] [CrossRef]
- Nazli, S.A.; Loeser, R.F.; Chubinskaya, S.; Willey, J.S.; Yammani, R.R. High fat-diet and saturated fatty acid palmitate inhibits IGF-1 function in chondrocytes. Osteoarthr. Cartil. 2017, 25, 1516–1521. [Google Scholar] [CrossRef]
- Alvarez-Garcia, O.; Rogers, N.H.; Smith, R.G.; Lotz, M.K. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 2014, 66, 1779–1788. [Google Scholar] [CrossRef]
- Bastiaansen-Jenniskens, Y.M.; Siawash, M.; Van de Lest, C.H.A.; Verhaar, J.A.N.; Kloppenburg, M.; Zuurmond, A.M.; Stojanovic-Susulic, V.; Van Osch, G.J.V.M.; Clockaerts, S. Monounsaturated and saturated, but not n-6 polyunsaturated fatty acids decrease cartilage destruction under inflammatory conditions: A preliminary study. Cartilage 2013, 4, 321–328. [Google Scholar] [CrossRef]
- Frommer, K.W.; Schäffler, A.; Rehart, S.; Lehr, A.; Müller-Ladner, U.; Neumann, E. Free fatty acids: Potential proinflammatory mediators in rheumatic diseases. Ann. Rheum. Dis. 2015, 74, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.C.; Goncalves, J.; Judas, F.; Mobasheri, A.; Lopes, C.; Mendes, A.F. Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res. Ther. 2009, 11, R80. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.C.; Rufino, A.T.; Judas, F.M.; Tenreiro, C.M.; Lopes, M.C.; Mendes, A.F. Role of glucose as a modulator of anabolic and catabolic gene expression in normal and osteoarthritic human chondrocytes. J. Cell. Biochem. 2011, 112, 2813–2824. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chan, D.C.; Lan, K.C.; Wang, C.C.; Chen, C.M.; Chao, S.C.; Tsai, K.S.; Yang, R.S.; Liu, S.H. PPARγ is involved in the hyperglycemia-induced inflammatory responses and collagen degradation in human chondrocytes and diabetic mouse cartilages. J. Orthop. Res. 2014, 33, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Laiguillon, M.C.; Courties, A.; Houard, X.; Auclair, M.; Sautet, A.; Capeau, J.; Fève, B.; Berenbaum, F.; Sellam, J. Characterization of diabetic osteoarthritic cartilage and role of high glucose environment on chondrocyte activation: Toward pathophysiological delineation of diabetes mellitus-related osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1513–1522. [Google Scholar] [CrossRef]
- Kelley, K.M.; Johnson, T.R.; Ilan, J.; Moskowitz, R.W. Glucose regulation of the IGF response system in chondrocytes: Induction of an IGF-I-resistant state. Am. J. Physiol. 1999, 276, 1164–1171. [Google Scholar] [CrossRef]
- Kimmerling, K.A.; Oswald, S.J.; Huebner, J.L.; Little, D.; Kraus, V.B.; Kang, J.X.; Wu, C.L.; Guilak, F. Transgenic conversion of ω-6 to ω-3 polyunsaturated fatty acids via fat-1 reduces the severity of post-traumatic osteoarthritis. Arthritis Res. Ther. 2020, 22, 83. [Google Scholar] [CrossRef]
- Tsubosaka, M.; Kihara, S.; Hayashi, S.; Nagata, J.; Kuwahara, T.; Fujita, M.; Kikuchi, K.; Takashima, Y.; Kamenaga, T.; Kuroda, Y.; et al. Gelatin hydrogels with eicosapentaenoic acid can prevent osteoarthritis progression in vivo in a mouse model. J. Orthop. Res. 2020, 38, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Zainal, Z.; Longman, A.J.; Hurst, S.; Duggan, K.; Caterson, B.; Hughes, C.E.; Harwood, J.L. Relative efficacies of omega-3 polyunsaturated fatty acids in reducing expression of key proteins in a model system for studying osteoarthritis. Osteoarthr. Cartil. 2009, 17, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Wann, A.K.T.; Mistry, J.; Blain, E.J.; Michael-Titus, A.T.; Knight, M.M. Eicosapentaenoic acid and docosahexaenoic acid reduce interleukin-1β-mediated cartilage degradation. Arthritis Res. Ther. 2010, 12, R207. [Google Scholar] [CrossRef] [PubMed]
- Adler, N.; Schoeniger, A.; Fuhrmann, H. Polyunsaturated fatty acids influence inflammatory markers in a cellular model for canine osteoarthritis. J. Anim. Physiol. Anim. Nutr. 2018, 102, e623–e632. [Google Scholar] [CrossRef]
- James, M.J.; Gibson, R.A.; Cleland, L.G. Dietary polyunsaturated fatty acids and inflammatory mediator production. Am. J. Clin. Nutr. 2000, 71, 343S–348S. [Google Scholar] [CrossRef] [PubMed]
- Martindale, R.G.; Warren, M.M.; McClave, S.A. Does the use of specialized proresolving molecules in critical care offer a more focused approach to controlling inflammation than that of fish oils? Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Monfoulet, L.E.; Philippe, C.; Mercier, S.; Coxam, V.; Wittrant, Y. Deficiency of G-protein coupled receptor 40, a lipid-activated receptor, heightens in vitro- and in vivo-induced murine osteoarthritis. Exp. Biol. Med. 2015, 240, 854–866. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, D.; Ho, K.W.; Lin, S.; Suen, W.C.W.; Zhang, H.; Zha, Z.; Li, G.; Leung, P.S. GPR120 is an important inflammatory regulator in the development of osteoarthritis. Arthritis Res. Ther. 2018, 20, 163. [Google Scholar] [CrossRef]
- Xu, Z.; Ke, T.; Zhang, Y.; Fu, C.; He, W. Agonism of GPR120 prevented IL-1β-induced reduction of extracellular matrix through SOX-9. Aging 2020, 12, 12074–12085. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.D.; Bertrand, G.; Vivot, K.; Carpentier, E.; Tremblay, C.; Ghislain, J.; Bouvier, M.; Poitout, V. β-arrestin recruitment and biased agonism at free fatty acid receptor 1. J. Biol. Chem. 2015, 290, 21131–21140. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Xiao, P.; Tao, X.N.; Qin, J.; He, Q.T.; Zhang, C.; Guo, S.C.; Du, Y.Q.; Chen, L.N.; Shen, D.D.; et al. Unsaturated bond recognition lead to biased signal in a fatty acid receptor. Science 2023, 380, eadd6220. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lin, H.; Zhang, Y.; Xu, T.; Wang, T.; Xue, X.; Zhang, W.; Liu, H. Activation of GPR40 supresses AGE-induced reduction in type II collagen and aggrecan in human SW1353 chondrocytes. Drug Des. Dev. Ther. 2020, 14, 2371–2379. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Sequences 5′-3′ |
|---|---|
| Aggrecan | Fwd: CAA-CCT-CCT-GGG-TGT-AAG-GA |
| Rev: TGT-AGC-AGA-TGG-CGT-CGT-AG | |
| Collagen 2 | Fwd: TCC-CTC-TGG-TTC-TGA-TGG-TC |
| Rev: CTC-TGT-CTC-CAG-ATG-CAC-CA | |
| COX-2 | Fwd: TAC-AAG-CAG-TGG-CAA-AGG-CC |
| Rev: CAG-TAT-TGA-GGA-GAA-CAG-ATG-GG | |
| mPGES | Fwd: ACC-CTC-TCA-TCG-CCT-GGA-TA |
| Rev: ATG-CGT-GGG-TTC-ATT-TTG-CC | |
| MMP-3 | Fwd: TCT-GGG-CTA-TCC-GAG-GTC-AT |
| Rev: TGC-ATC-GAT-CTT-CTG-GAC-GG | |
| MMP-13 | Fwd: TCT-GGG-CTA-TCC-GAG-GTC-AT |
| Rev: TGC-ATC-GAT-CTT-CTG-GAC-GG | |
| RP29 | Fwd: CTC-TAA-CCG-CCA-CGG-TCT-GA |
| Rev: ACT-AGC-ATG-ATT-GGT-ATC-AC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, C.; Presle, N.; Pizard, A.; Guillaume, C.; Bianchi, A.; Kempf, H. Beneficial Impact of Eicosapentaenoic Acid on the Adverse Effects Induced by Palmitate and Hyperglycemia on Healthy Rat Chondrocyte. Int. J. Mol. Sci. 2024, 25, 1810. https://doi.org/10.3390/ijms25031810
Deng C, Presle N, Pizard A, Guillaume C, Bianchi A, Kempf H. Beneficial Impact of Eicosapentaenoic Acid on the Adverse Effects Induced by Palmitate and Hyperglycemia on Healthy Rat Chondrocyte. International Journal of Molecular Sciences. 2024; 25(3):1810. https://doi.org/10.3390/ijms25031810
Chicago/Turabian StyleDeng, Chaohua, Nathalie Presle, Anne Pizard, Cécile Guillaume, Arnaud Bianchi, and Hervé Kempf. 2024. "Beneficial Impact of Eicosapentaenoic Acid on the Adverse Effects Induced by Palmitate and Hyperglycemia on Healthy Rat Chondrocyte" International Journal of Molecular Sciences 25, no. 3: 1810. https://doi.org/10.3390/ijms25031810