

T-Cell Receptor Sequences Identify Combined Coxsackievirus–Streptococci Infections as Triggers for Autoimmune Myocarditis and Coxsackievirus–Clostridia Infections for Type 1 Diabetes

Abstract
:1. Introduction
Problems and Hypotheses
2. Results
3. Discussion
3.1. Summary
3.2. Further Evidence of Both Clostridia and Coxsackieviruses in T1DM
3.3. Further Evidence of Both Coxsackieviruses and Streptococci in AM
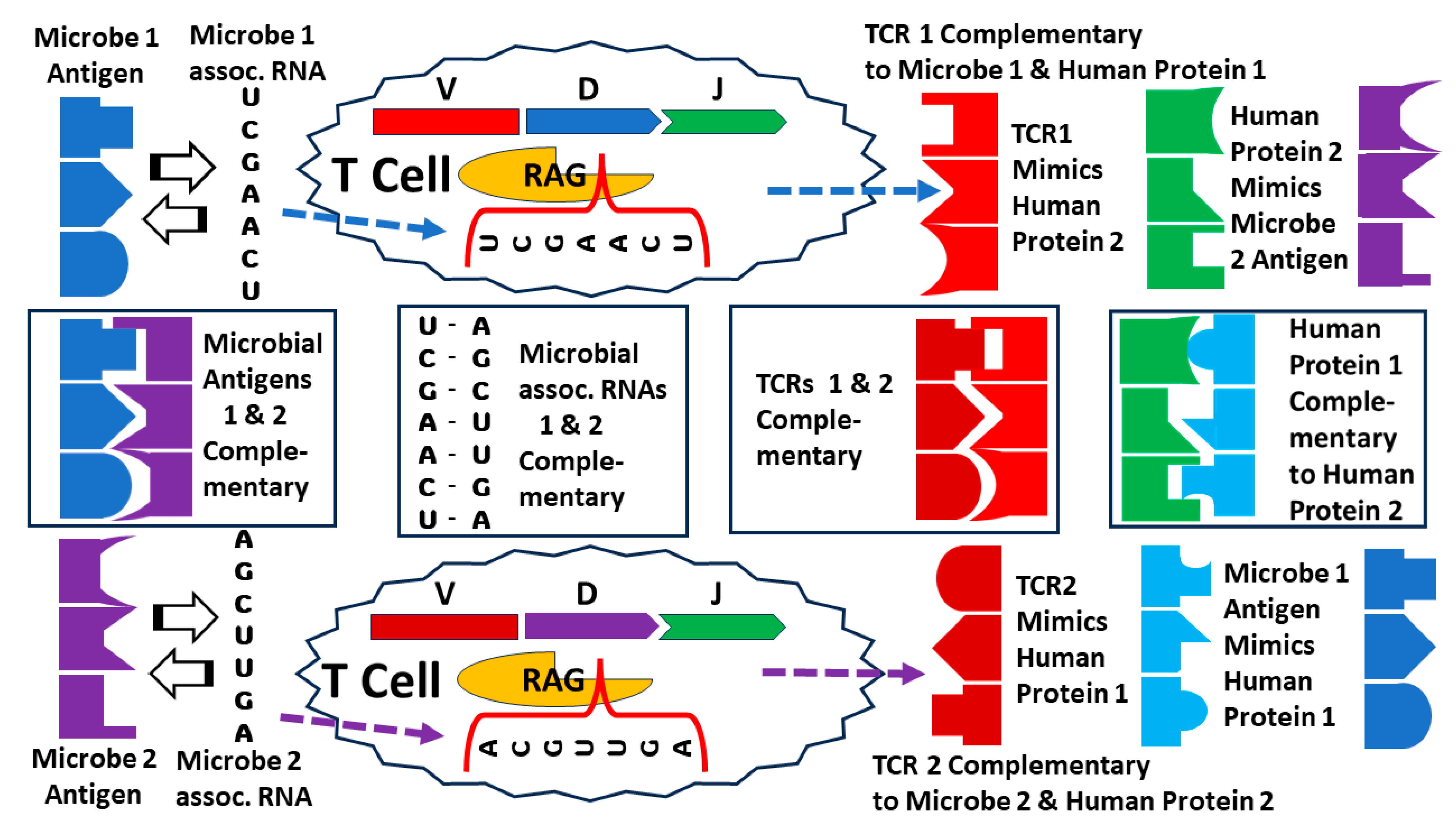
3.4. Explaining the Results, 1: Molecular Mimicry Theory
3.5. Explaining the Results, 2: Bystander Activation
3.6. Explaining the Results, 3: The Anti-Idiotype Theory
3.7. Explaining the Results, 4: Complementary Antigen Theory
3.8. Explaining the Results, 5: Antigen Templating of the Hypervariable Region
3.9. Experimental Tests to Differentiate the Various Theories
3.10. Elucidating AD Etiologies from TCR Sequences to Develop Novel Animal Models
3.11. Implications for Loss of Microbiome Tolerance in Autoimmune Diseases
3.12. Implications for Coxsackievirus and Clostridia Vaccine Development
3.13. Limitations of the Study
4. Materials and Methods
4.1. Similarity Searches
4.2. TCR Sources
4.3. Statistics
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

| Human Pathogen | NOR TCR % N = 325 | T1D TCR % N = 79 | MYO TCR % N = 34 | p-Value (X2) T1D vs. NOR | p-Value (X2) MYO vs. NOR | p-Value (X2) MYO vs. T1D |
|---|---|---|---|---|---|---|
| Adenovirus | 14 | 15 | 3 | (0.003) | (0.0001) | |
| Astrovirus | 2 | 0 | 3 | |||
| Bocavirus | 0 | 1 | 0 | |||
| Cardiovirus | 0 | 1 | 0 | |||
| Coronavirus | 3 | 1 | 0 | |||
| Coxsackie A | 5 | 5 | 15 | 0.004 | 0.004 | |
| Coxsackie B | 3 | 34 | 30 | <0.0001 | <0.0001 | |
| CMV | 21 | 26 | 12 | |||
| Echoviruses | 10 | 3 | 3 | (<0.005) | (<0.005) | |
| Enteroviruses | 10 | 3 | 6 | |||
| EBV | 5 | 10 | 0 | |||
| HAV | 1 | 0 | 0 | |||
| HBV | 12 | 10 | 9 | |||
| HCV | 18 | 24 | 3 | (<0.0001) | ||
| HEV | 1 | 3 | 3 | |||
| HHV1 | 5 | 4 | 9 | |||
| HHV2 | 3 | 9 | 0 | |||
| HHV6 | 6 | 1 | 0 | |||
| HHV8 | 2 | 3 | 9 | |||
| HIV-1 | 74 | 69 | 53 | |||
| HTLV | 1 | 0 | 3 | |||
| Infl A Virus | 24 | 24 | 9 | (<0.0001) | ||
| Infl B virus | 1 | 1 | 3 | |||
| Infl C virus | 0 | 1 | 0 | |||
| Jap enc virus | 2 | 1 | 0 | |||
| Measles virus | 6 | 0 | 3 | |||
| Mumps virus | 1 | 0 | 0 | |||
| Norovirus | 8 | 6 | 3 | |||
| Papilloma virus | 35 | 12 | 15 | (<0.0001) | (<0.0001) | |
| Parainfluenza | 1 | 0 | 0 | |||
| Polio virus | 0 | 0 | 0 | |||
| Polyoma virus | 2 | 1 | 3 | |||
| Reovirus | 10 | 1 | 3 | |||
| RSV | 0 | 0 | 3 | |||
| Rhinovirus | 3 | 6 | 0 | |||
| Rotaviruses | 9 | 4 | 3 | |||
| Rubella | 2 | 3 | 0 | |||
| Varicella zoster | 3 | 3 | 3 |
| Phages | NOR TCR % N = 104 | T1D TCR % N = 68 | p-Value T1D vs. NOR |
|---|---|---|---|
| Bacillus | 41 | 15 | (<0.0001) |
| Bacteroides | 0 | 0 | |
| Bifidobacteria | 0 | 0 | |
| Campylobacter | 9 | 6 | |
| Chlamydia | 2 | 0 | |
| Clostridium | 15 | 9 | |
| Corynebacterium | 2 | 0 | |
| Enterobacterium | 53 | 25 | (<0.0001) |
| Enterococcus | 13 | 2 | |
| Escherichia | 46 | 24 | (<0.0001) |
| Giardia lamblia | 0 | 0 | |
| Haemophilus | 2 | 0 | |
| Klebsiella | 15 | 10 | |
| Lactobacillus | 20 | 6 | |
| Lactococcus | 11 | 10 | |
| Listeria | 5 | 2 | |
| Mycobacterium | 61 | 37 | |
| Mycoplasma | 0 | 2 | |
| Pseudomonas | 38 | 34 | |
| Salmonella | 34 | 10 | |
| Serratia | 2 | 3 | |
| Shigella | 17 | 9 | |
| Staphylococcus | 18 | 13 | |
| Streptococcus | 13 | 6 | |
| T. vaginitis | 0 | 0 |
| Human Pathogen | NOR TCR % N = 325 | T1D TCR % N = 79 | MYO TCR % N = 34 | p-Value (X2) NOR vs. T1D | p-Value (X2) NOR vs. MYO | p-Value (X2) MYO vs. T1D |
|---|---|---|---|---|---|---|
| Bacillus cereus | 24 | 23 | 9 | |||
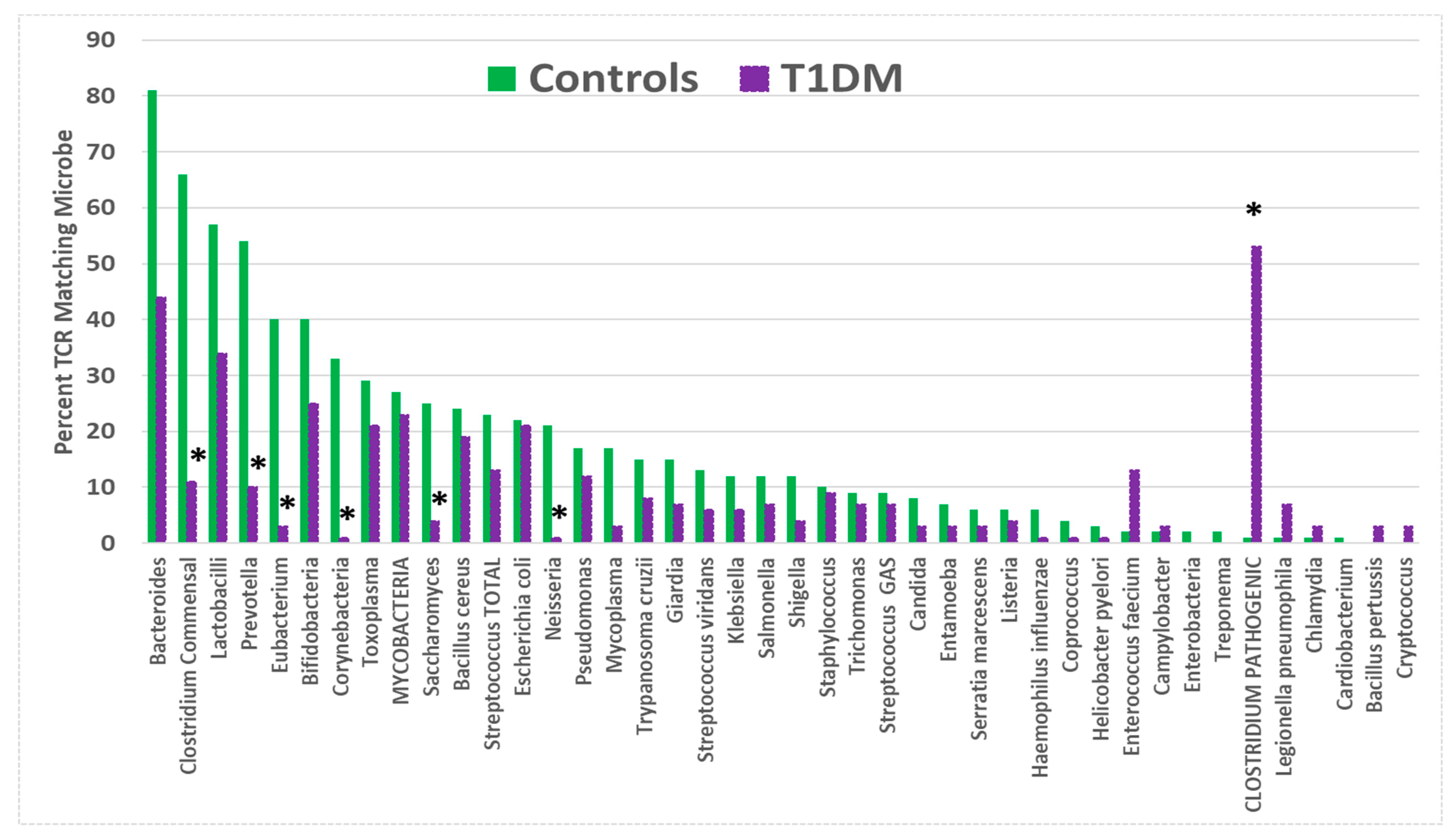
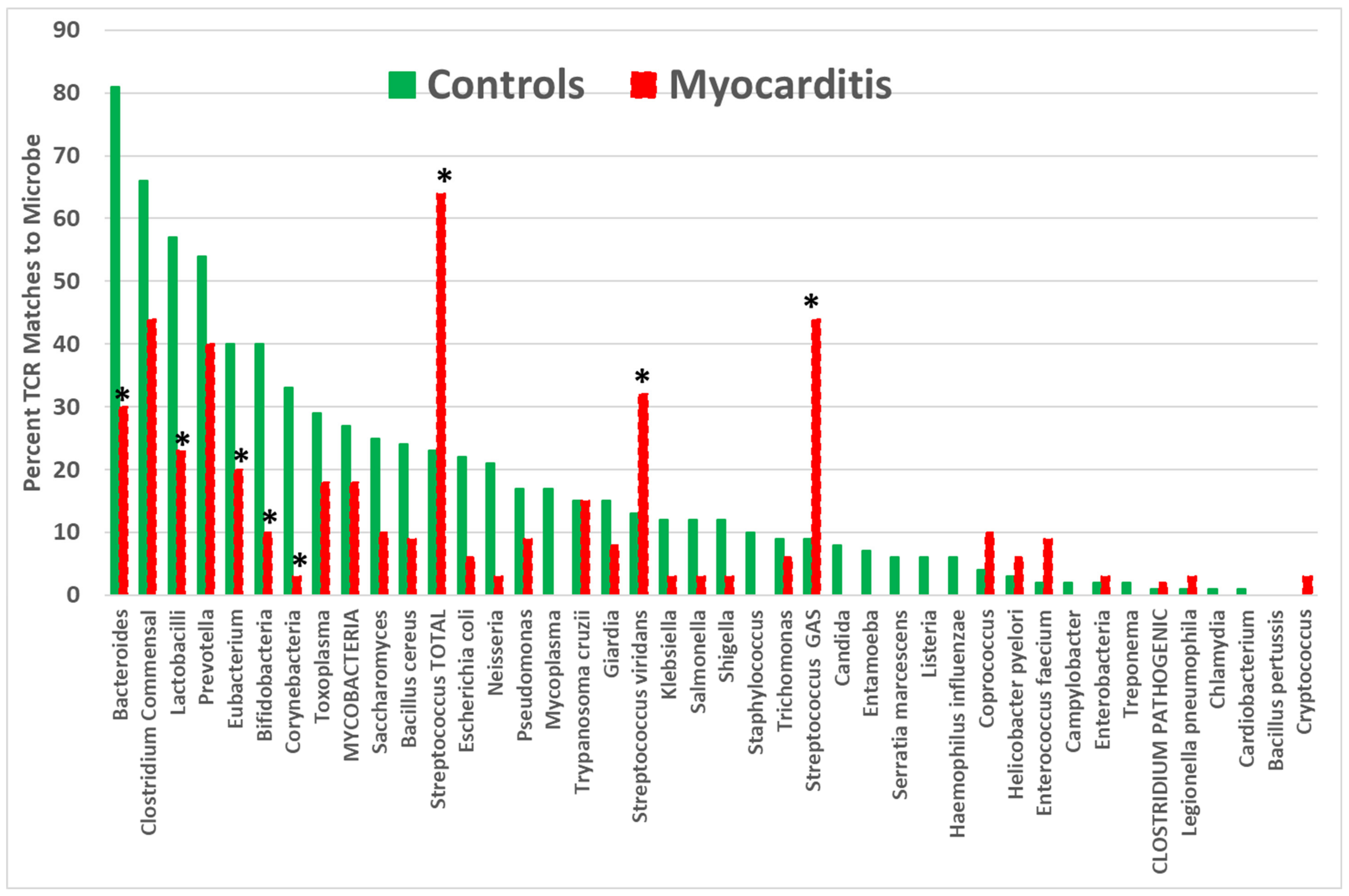
| Bacteroides | 81 | 46 | 30 | (<0.0001) | (<0.0001) | |
| Bifidobacteria | 40 | 23 | 10 | |||
| B. pertussis | 1 | 3 | 0 | |||
| Campylobacter jejuni | 2 | 3 | 0 | |||
| Cardiobacterium | 1 | 0 | 0 | |||
| Chlamydia pneumoniae | 2 | 3 | 0 | |||
| CLOSTRIDIUM PATHOGENIC. | 9 | 55 | 12 | <0.0001 | (<0.0001) | |
| Clostridium commensal | 66 | 13 | 44 | (<0.0001) | (<0.0001) | |
| Coprococcus | 4 | 6 | 3 | |||
| Cornybacterium | 6 | 1 | 3 | |||
| Enterobacter sp. | 2 | 0 | 3 | |||
| Enterococcus faecium | 28 | 12 | 9 | |||
| Escherichia coli | 22 | 27 | 6 | (<0.0001) | ||
| Eubacterium | 40 | 4 | 20 | (<0.0001) | (<0.0001) | <0.0001 |
| Haemophilus influenzae | 6 | 1 | 0 | |||
| Helicobacter pyelori | 3 | 1 | 6 | (0.002) | ||
| Klebsiella pneumoniae | 12 | 6 | 3 | |||
| Lactobacilli | 57 | 37 | 23 | (0.002) | ||
| Legionella pneumophila | 3 | 6 | 3 | |||
| Listeria | 6 | 4 | 0 | |||
| M. tuberculosis | 2 | 7 | 3 | |||
| Mycobacteria (atypical) | 25 | 16 | 12 | |||
| Mycobacteria Tot. | 27 | 26 | 18 | |||
| Mycoplasma sp. | 7 | 3 | 0 | |||
| Neisseria sp. | 9 | 1 | 3 | |||
| Prevotella | 54 | 11 | 41 | (<0.0001) | ||
| Pseudomonas aeruginosa | 17 | 12 | 9 | |||
| Salmonella sp. | 18 | 7 | 3 | |||
| Serratia marcescens | 6 | 3 | 0 | |||
| Shigella dysenteriae | 3 | 4 | 3 | |||
| Staphylococcus sp. | 10 | 9 | 0 | |||
| STREPTOCOCCUS GAS | 23 | 7 | 44 | <0.0001 | <0.0001 | |
| STREPTOCOCCUS VIR | 17 | 6 | 32 | <0.0001 | ||
| STREP. TOTAL | 29 | 13 | 64 | <0.0001 | <0.0001 | |
| Trichomonas vaginalis | 15 | 7 | 6 |
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Appendix C

References
- Root-Bernstein, R. Autoreactive T-cell receptor (Vbeta/D/Jbeta) sequences in diabetes are homologous to insulin, glucagon, the insulin receptor, and the glucagon receptor. J. Mol. Recognit. 2009, 22, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Autoimmunity and the microbiome: T-cell receptor mimicry of “self” and microbial antigens mediates self tolerance in holobionts. BioEssays 2016, 38, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Beseme, S.; Tassone, R.; Liu, R.; Kibria, F.; Terry, F.; Martin, W.; De Groot, A.S. T cell epitope redundancy: Cross-conservation of the TCR face between pathogens and self and its implications for vaccines and autoimmunity. Expert Rev. Vaccines 2016, 15, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Terry, F.; Gutierrez, A.H.; Tassone, R.; Losikoff, P.; Gregory, S.H.; Bailey-Kellogg, C.; Martin, W.D.; De Groot, A.S. Smarter vaccine design will circumvent regulatory T cell-mediated evasion in chronic HIV and HCV infection. Front. Microbiol. 2016, 5, 502. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.I. A hypothesis: Establishing the microbiome through immune mimicry. Bioessays 2016, 38, 1062. [Google Scholar] [CrossRef] [PubMed]
- Swiatczak, B.; Tauber, A.I. Holoimmunity Revisited. Bioessays 2018, 40, e1800117. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Human Immunodeficiency Virus Proteins Mimic Human T Cell Receptors Inducing Cross-Reactive Antibodies. Int. J. Mol. Sci. 2017, 18, 2091. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Churchill, E.; Oliverio, S. T Cell Receptor Sequences Amplified during Severe COVID-19 and Multisystem Inflammatory Syndrome in Children Mimic SARS-CoV-2, Its Bacterial Co-Infections and Host Autoantigens. Int. J. Mol. Sci. 2023, 24, 1335. [Google Scholar] [CrossRef]
- Caforio, A.L.; Iliceto, S. Genetically determined myocarditis: Clinical presentation and immunological characteristics. Curr. Opin. Cardiol. 2008, 23, 219–226. [Google Scholar] [CrossRef]
- Li, H.S.; Ligons, D.L.; Rose, N.R. Genetic complexity of autoimmune myocarditis. Autoimmun. Rev. 2008, 7, 168–173. [Google Scholar] [CrossRef]
- Van Belle, T.L.; Coppieters, K.T.; von Herrath, M.G. Type 1 diabetes: Etiology, immunology, and therapeutic strategies. Physiol. Rev. 2011, 91, 79–118. [Google Scholar] [CrossRef]
- Pugliese, A. The multiple origins of Type 1 diabetes. Diabet. Med. 2013, 30, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, L.; Köhler, K.F.; Postol, E.; Kalil, J. Genes, autoimmunity and pathogenesis of rheumatic heart disease. Ann. Pediatr. Cardiol. 2011, 4, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Ihm, S.H.; Kim, K.W. Viruses as a triggering factor of type 1 diabetes and genetic markers related to the susceptibility to the virus-associated diabetes. Diabetes Res. Clin. Pract. 1989, 7 (Suppl. S1), S47–S58. [Google Scholar] [CrossRef] [PubMed]
- Hober, D.; Sauter, P. Pathogenesis of type 1 diabetes mellitus: Interplay between enterovirus and host. Nat. Rev. Endocrinol. 2010, 6, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.J.; Horwitz, M.S. Coxsackievirus infection as an environmental factor in the etiology of type 1 diabetes. Autoimmun. Rev. 2009, 8, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Jaidane, H.; Hober, D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008, 34, 537–548. [Google Scholar] [CrossRef]
- Nair, A.; Wolter, T.R.; Meyers, A.J.; Zipris, D. Innate immune pathways in virus-induced autoimmune diabetes. Ann. N. Y. Acad. Sci. 2008, 1150, 139–142. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Alidjinou, E.K.; Hober, D. Persistent coxsackievirus B infection and patho-genesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2022, 18, 503–516. [Google Scholar] [CrossRef]
- Roivainen, M.; Knip, M.; Hyöty, H.; Kulmala, P.; Hiltunen, M.; Vähäsalo, P.; Hovi, T.; Akerblom, H.K. Several different enterovirus serotypes can be associated with prediabetic autoimmune episodes and onset of overt IDDM. Child. Diabetes Finl. (DiMe) Study Group J. Med. Virol. 1998, 56, 74–78. [Google Scholar] [CrossRef]
- Sané, F.; Moumna, I.; Hober, D. Group B coxsackieviruses and autoimmunity: Focus on Type 1 diabetes. Expert. Rev. Clin. Immunol. 2011, 7, 357–366. [Google Scholar] [CrossRef]
- Goldberg, E.; Krause, I. Infection and type 1 diabetes mellitus—A two edged sword? Autoimmun. Rev. 2009, 8, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Lammi, N.; Karvonen, M.; Tuomilehto, J. Do microbes have a causal role in type 1 diabetes? Med. Sci. Monit. 2005, 11, RA63-9. [Google Scholar] [PubMed]
- Jun, H.S.; Yoon, J.W. A new look at viruses in type 1 diabetes. Diabetes Metab. Res. Rev. 2003, 19, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Fromantin, M.; Laverdant, C. Le diabète de l’hépatite virale: À propos de vingt nouvelles observations [Diabetes following viral hepatitis: 20 recent cases]. Diabete 1970, 18, 121–125. (In French) [Google Scholar] [PubMed]
- Franczak, T. Cukrzyca w nastepstwie wirusowego zapalenia watroby [Diabetes following virus hepatitis]. Pol. Tyg. Lek. 1969, 24, 1705–1706. (In Polish) [Google Scholar] [PubMed]
- Rosu, V.; Ahmed, N.; Paccagnini, D.; Gerlach, G.; Fadda, G.; Hasnain, S.E.; Zanetti, S.; Sechi, L.A. Specific immunoassays confirm association of Mycobacterium avium Subsp. paratuberculosis with Type-1 but not Type-2 diabetes mellitus. PLoS ONE 2009, 4, e4386. [Google Scholar] [CrossRef]
- Sechi, L.A.; Rosu, V.; Pacifico, A.; Fadda, G.; Ahmed, N.; Zanetti, S. Humoral immune responses of type 1 diabetes patients to Mycobacterium avium subsp. paratuberculosis lend support to the infectious trigger hypothesis. Clin. Vaccine Immunol. 2008, 15, 320–326. [Google Scholar] [CrossRef]
- Satorres, S.; Alcaraz, L.; Di Genaro, S. Association between high levels IL-8 and Staphylococcus aureus-specific IgA antibodies in subjects with type 1 diabetes mellitus from Argentina. Diabetes Res. Clin. Pract. 2007, 77, 489–491. [Google Scholar] [CrossRef]
- Neophytou, P.I.; Roep, B.O.; Arden, S.D.; Muir, E.M.; Duinkerken, G.; Kallan, A.; de Vries, R.R.; Hutton, J.C. T-cell epitope analysis using subtracted expression libraries (TEASEL): Application to a 38-kDA autoantigen recognized by T cells from an insulin-dependent diabetic patient. Proc. Natl. Acad. Sci. USA 1996, 93, 2014–2018. [Google Scholar] [CrossRef]
- Marietti, M.; Gasbarrini, A.; Saracco, G.; Pellicano, R. Helicobacter pylori infection and diabetes mellitus: The 2013 state of art. Panminerva Med. 2013, 55, 277–281. [Google Scholar] [PubMed]
- Abdellatif, A.M.; Jensen Smith, H.; Harms, R.Z.; Sarvetnick, N.E. Human Islet Response to Selected Type 1 Diabetes-Associated Bacteria: A Transcriptome-Based Study. Front. Immunol. 2019, 10, 2623. [Google Scholar] [CrossRef] [PubMed]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyöty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Goffau, M.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 610, e25792. [Google Scholar] [CrossRef] [PubMed]
- Murri, M.; Leiva, L.; Gomez-Zumaquero, J.M.; Tinahones, F.J.; Cardona, F. Gut microbiota in children with type 1 diabetes differs from that in healthy children: A case-control study. BMC Med. 2013, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, O. Human intestinal microbiota and Type 1 Diabetes. Curr. Diab. Rep. 2013, 13, 601–607. [Google Scholar] [CrossRef]
- Von Herrath, M.G.; Holz, A.; Homann, D.; Oldstone, M.B. Role of viruses in type I diabetes. Semin. Immunol. 1998, 10, 87–100. [Google Scholar] [CrossRef]
- Zipris, D. Epidemiology of type 1 diabetes and what animal models teach us about the role of viruses in disease mechanisms. Clin. Immunol. 2009, 131, 11–23. [Google Scholar] [CrossRef]
- Babu, P.G.; John, T.J. Alteration of immune response to coxsackie B3 virus by streptozotocin in dual-aetiology diabetes mellitus in mouse. Indian J. Exp. Biol. 1987, 25, 17–21. [Google Scholar]
- Yoon, J.W.; London, W.T.; Curfman, B.L.; Brown, R.L.; Notkins, A.L. Coxsackie virus B4 produces transient diabetes in nonhuman primates. Diabetes 1986, 35, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Taylor, G.; Basid, A. The effect of pertussis vaccine on the insulin-dependent diabetes induced by streptozotocin in ice. Pediatr. Res. 1984, 18, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Nomaguchi, H.; Yogi, Y.; Kawatsu, K.; Okamura HOzawa, Y.; Kasatani, T. Prevention of diabetes in non-obese diabetic mice by a single immunization with Mycobacterium leprae. Nihon Hansen. Gakkai Zasshi 2002, 71, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Silveira, P.A.; Baxter, A.G. The NOD mouse as a model of SLE. Autoimmunity 2001, 34, 53–64. [Google Scholar] [CrossRef]
- Kühtreiber, W.M.; Faustman, D.L. BCG Therapy for Type 1 Diabetes: Restoration of Balanced Immunity and Metabolism. Trends Endocrinol. Metab. 2019, 30, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, M.S.; Bradley, L.M.; Harbertson, J.; Krahl, T.; Lee, J.; Sarvetnick, N. Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat. Med. 1998, 4, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.; von Herrath, M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell. Immunol. 2005, 233, 125–132. [Google Scholar] [CrossRef]
- Rose, N.R.; Hill, S.L. The pathogenesis of postinfectious myocarditis. Clin. Immunol. Immunopathol. 1996, 80, S92–S99. [Google Scholar] [CrossRef]
- Ciháková, D.; Rose, N.R. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv. Immunol. 2008, 99, 95–114. [Google Scholar] [CrossRef]
- Abrams, D.; Derrick, G.; Penny, D.J.; Shinebourne, E.A.; Redington, A.N. Cardiac complications in children following infection with varicella zoster virus. Cardiol. Young. 2001, 11, 647–652. [Google Scholar] [CrossRef]
- Bowles, N.E.; Ni, J.; Kearney, D.L.; Pauschinger, M.; Schultheiss, H.P.; McCarthy, R.; Hare, J.; Bricker, J.T.; Bowles, K.R.; Towbin, J.A. Detection of viruses in myocardial tissues by polymerase chain reaction: Evidence of adenovirus as a common cause of myocarditis in children and adults. J. Am. Coll. Cardiol. 2003, 42, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yamada, T.; Matsumori, A. Hepatitis C virus and cardiomyopathy. In Cardiomyopathies and Heart Failure: Biomolecular, Infectious, and Immune Mechanisms; Matsumori, A., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 2003; pp. 325–339. [Google Scholar]
- Pauschinger, M.; Bowles, N.E.; Fuentes-Garcia, F.J.; Pham, V.; Kuhl, U.; Schwimmbeck, P.L.; Schultheiss, H.P.; Towbin, J.A. Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation 1999, 99, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Panhoweit, S.; Lamparter, S.; Schoppet, M.; Maisch, B. Parvovirus B19 genome in endomyocardial biopsy specimens. Circulation 2004, 109, e179. [Google Scholar]
- Mahfoud, F.; Gärtner, B.; Kindermann, M.; Ukena, C.; Gadomski, K.; Klingel, K.; Kandolf, R.; Böhm, M.; Kindermann, I. Virus serology in patients with suspected myocarditis: Utility or futility? Eur. Heart J. 2011, 32, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Andréoletti, L.; Lévêque, N.; Boulagnon, C.; Brasselet, C.; Fornes, P. Viral causes of human myocarditis. Arch. Cardiovasc. Dis. 2009, 102, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Halsell, J.S.; Riddle, J.R.; Atwood, J.E.; Gardner, P.; Shope, R.; Poland, G.A.; Gray, G.C.; Ostroff, S.; Eckart, R.E.; Hospenthal, D.R.; et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. JAMA 2003, 289, 3283–3289. [Google Scholar] [CrossRef] [PubMed]
- Henao-Martínez, A.F.; Schwartz, D.A.; Yang, I.V. Chagasic cardiomyopathy, from acute to chronic: Is this mediated by host susceptibility factors? Trans. R. Soc. Trop. Med. Hyg. 2012, 106, 521–527. [Google Scholar] [CrossRef]
- Cunningham, M.W. Streptococcus and rheumatic fever. Curr. Opin. Rheumatol. 2012, 24, 408–416. [Google Scholar] [CrossRef]
- Guilherme, L.; Kalil, J. Rheumatic fever: From sore throat to autoimmune heart lesions. Int. Arch. Allergy Immunol. 2004, 134, 56–64. [Google Scholar] [CrossRef]
- Sikder, S.; Williams, N.L.; Sorenson, A.E.; Alim, M.A.; Vidgen, M.E.; Moreland, N.J.; Rush, C.M.; Simpson, R.S.; Govan, B.L.; Norton, R.E.; et al. Group G Streptococcus Induces an Autoimmune Carditis Mediated by Interleukin 17A and Interferon γ in the Lewis Rat Model of Rheumatic Heart Disease. J. Infect. Dis. 2018, 218, 324–335. [Google Scholar] [CrossRef]
- Makaryus, A.N.; Revere, D.J.; Steinberg, B. Recurrent reversible dilated cardiomyopathy secondary to viral and streptococcal pneumonia vaccine-associated myocarditis. Cardiol. Rev. 2006, 14, e1–e4. [Google Scholar] [CrossRef]
- Stewart, G.C.; Lopez-Molina, J.; Gottumukkala, R.V.; Rosner, G.F.; Anello, M.S.; Hecht, J.L.; Winters, G.L.; Padera, R.F.; Baughman, K.L.; Lipes, M.A. Myocardial parvovirus B19 persistence: Lack of association with clinicopathologic phenotype in adults with heart failure. Circ. Heart Fail. 2011, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Poloniecki, L.A.; Caforio, A.L.; Davies, M.J.; Booth, J.C.; McKenna, W.J. A prospective case-control study of antibodies to Coxsackie B virus in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 1994, 23, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Ciháková, D.; Sharma, R.B.; Fairweather, D.; Afanasyeva, M.; Rose, N.R. Animal models for autoimmune myocarditis and autoimmune thyroiditis. Methods Mol. Med. 2004, 102, 175–193. [Google Scholar] [PubMed]
- Fairweather, D.; Stafford, K.A.; Sung, Y.K. Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 2012, 24, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.M.; Fairweather, D.; Huber, S.A.; Cunningham, M.W. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr. Protoc. Immunol. 2013, 101, 15.14.1–15.14.51. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.M.; O’Donoghue, H.L.; Reed, W.D. Mouse cytomegalovirus infection induces antibodies which cross-react with virus and cardiac myosin: A model for the study of molecular mimicry in the pathogenesis of viral myocarditis. Immunology 1992, 75, 513–519. [Google Scholar] [PubMed]
- Masedunskas, A.; Porat-Shliom, N.; Weigert, R. Linking differences in membrane tension with the requirement for a contractile actomyosin scaffold during exocytosis in salivary glands. Commun. Integr. Biol. 2012, 5, 84–87. [Google Scholar] [CrossRef]
- Gorton, D.; Blyth, S.; Gorton, J.G.; Govan, B.; Ketheesan, N. An alternative technique for the induction of autoimmune valvulitis in a rat model of rheumatic heart disease. J. Immunol. Methods 2010, 355, 80–85. [Google Scholar] [CrossRef]
- Xie, X.; Zhou, H.; Huang, J.; Huang, H.; Feng, Z.; Mei, K.; Yu, B.; Su, Z.; Gu, J. An animal model of chronic rheumatic valvulitis induced by formalin-killed streptococci. Rheumatol. Int. 2010, 30, 1621–1625. [Google Scholar] [CrossRef]
- Burch, G.E.; Giles, T.D. The role of viruses in the production of heart disease. Am. J. Cardiol. 1972, 29, 231–240. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Vonck, J.; Podufaly, A. Antigenic complementarity between coxsackie virus and streptococci in rheumatic heart disease and myocarditis. Autoimmunity 2009, 22, 177–187. [Google Scholar]
- Dale, J.B.; Fischetti, V.A.; Carapetis, J.R.; Steer, A.C.; Sow, S.; Kumar, R.; Mayosi, B.M.; Rubin, F.A.; Mulholland, K.; Hombach, J.M.; et al. Group A streptococcal vaccines: Paving a path for accelerated development. Vaccine 2013, 31 (Suppl. S2), B216–B222. [Google Scholar] [CrossRef]
- Zaher, S.R.; Kassem, A.S.; Hughes, J.J. Coxsackie virus infections in rheumatic fever. Indian J. Pediatr. 1993, 60, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Suresh, L.; Chandrasekar, S.; Rao, R.S.; Ravi, V.; Badrinath, S. Coxsackie virus and rheumatic fever. A correlative study. J. Assoc. Physicians India 1989, 37, 582–585. [Google Scholar] [PubMed]
- Olgunturk, R.; Okur, I.; Cirak, M.Y.; Oguz, A.D.; Akalin, N.; Turet, S.; Tunaoglu, S. The role of viral agents in aetiopathogenesis of acute rheumatic fever. Clin Rheumatol. 2011, 30, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Vikerfors, T.; Stjerna, A.; Olcén, P.; Malmcrona, R.; Magnius, L. Acute myocarditis. Serologic diagnosis, clinical findings and follow-up. Acta Med. Scand. 1988, 223, 45–52. [Google Scholar] [CrossRef]
- Górska, A.; Urban, M.; Głowińska, B.; Kowalewski, M. Czy zakazenie paciorkowcem grupy A jest wyłaczna infekcyjna przyczyna goraczki reumatycznej?—Opis przebiegu goraczki reumatycznej u 11-letniego chłopca ze współistniejaca infekcja wirusem Coxackie B1 [Is infection with group A streptococcus the only reason for rheumatic fever?—A case report of rheumatic fever coexisting with Coxsackie B1 virus infection]. Przegl. Lek. 1998, 55, 418–419. (In Polish) [Google Scholar]
- Novikov, I. O diagnostike nerevmaticheskikh miokarditov [Diagnosis of nonrheumatic myocarditis]. Kardiologiia 1983, 23, 50–55. (In Russian) [Google Scholar]
- Cotor, F.; Zavate, O.; Finichiu, M.; Avram, G.; Ivan, A. Enterovirus contamination of swimming pool water; correlation with bacteriological indicators. Virologie 1983, 34, 251–256. [Google Scholar]
- Kogut, E.P.; Levashova, N.V.; Bondarenko, A.P.; Zherdeva, A.I.; Shuvalova, I.A. Eksperimental’noe izuchenie koksaki-streptokokkovoĭ infektsii [Experimental study of Coxsackie-streptococcal infection]. Vopr. Virusol. 1978, 690–695. (In Russian) [Google Scholar]
- Kuhl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheisss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, N.; Leveque, N.; Corcia, P.; Brunaud-Danel, V.; Salort-Campana, E.; Besson, G.; Tranchant, C.; Clavelou, P.; Beaulieux, F.; Ecochard, R.; et al. Cerebrospinal fluid detection of enterovirus genome in ALS: A study of 242 patients and 354 controls. Amyotroph. Lateral Scler. 2010, 11, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllopoulou, A.; Tapinos, N.; Moutsopoulos, H.M. Evidence for coxsackievirus infection in primary Sjögren’s syndrome. Arthritis Rheum 2004, 50, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulou, E.A.; Routsias, J.G.; Stea, E.A.; Moutsopoulos, H.M.; Tzioufas, A.G. Cross-reaction between antibodies to the major epitope of Ro60 kD autoantigen and a homologous peptide of coxsackie virus 2B protein. Clin. Exp. Immunol. 2005, 141, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Utomo, S.W.; Putri, J.F. Infections as Risk Factor of Sjögren’s Syndrome. Open Access Rheumatol. 2020, 12, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Olival, G.S.; Lima, B.M.; Sumita, L.M.; Serafim, V.; Fink, M.C.; Nali, L.H.; Romano, C.M.; Thomaz, R.B.; Cavenaghi, V.B.; Tilbery, C.P.; et al. Multiple sclerosis and herpesvirus interaction. Arq. Neuropsiquiatr. 2013, 71, 727–730. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, J.; Wei, X.; Wang, X.; Li, Y.; Peng, B.; Niu, T. The role of Epstein-Barr virus (EBV) and cytomegalovirus (CMV) in immune thrombocytopenia. Hematology 2013, 18, 295–299. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Couturier, J. Antigenic complementarity in the origins of autoimmunity: A general theory illustrated with a case study of idiopathic thrombocytopenia purpura. Clin. Dev. Immunol. 2006, 13, 49–65. [Google Scholar] [CrossRef]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus in systemic autoimmune diseases. Clin. Dev. Immunol. 2013, 2013, 535–538. [Google Scholar] [CrossRef]
- Kivity, S.; Arango, M.T.; Ehrenfeld, M.; Tehori, O.; Shoenfeld, Y.; Anaya, J.M.; Agmon-Levin, N. Infection and autoimmunity in Sjogren’s syndrome: A clinical study and comprehensive review. J. Autoimmun. 2014, 51, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Rozenblyum, E.V.; Allen, U.D.; Silverman, E.D.; Levy, D.M. Cytomegalovirus infection in childhood-onset systemic lupus erythematosus. Int. J. Clin. Rheumtol. 2013, 8, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Caudie, C.; Quittard Pinon, A.; Taravel, D.; Sivadon-Tardy, V.; Orlikowski, D.; Rozenberg, F.; Sharshar, T.; Raphaël, J.C.; Gaillard, J.L. Preceding infections and anti-ganglioside antibody profiles assessed by a dot immunoassay in 306 French Guillain-Barré syndrome patients. J. Neurol. 2011, 258, 1958–1964. [Google Scholar] [CrossRef] [PubMed]
- Lo Schiavo, A.; Ruocco, E.; Brancaccio, G.; Caccavale, S.; Ruocco, V.; Wolf, R. Bullous pemphigoid: Etiology, pathogenesis, and inducing factors: Facts and controversies. Clin. Dermatol. 2013, 31, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. Rethinking Molecular Mimicry in Rheumatic Heart Disease and Autoimmune Myocarditis: Laminin, Collagen IV, CAR, and B1AR as Initial Targets of Disease. Front. Pediatr. 2014, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Fairweather, D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J. Theor. Biol. 2015, 375, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Fairweather, D. Complexities in the relationship between infection and autoimmunity. Curr. Allergy Asthma Rep. 2014, 14, 407. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Chiles, K.; Huber, J.; Ziehl, A.; Turke, M.; Pietrowicz, M. Clostridia and Enteroviruses as Synergistic Triggers of Type 1 Diabetes Mellitus. Int. J. Mol. Sci. 2023, 24, 8336. [Google Scholar] [CrossRef]
- Liu, Y.; Batchuluun, B.; Ho, L.; Zhu, D.; Prentice, K.J.; Bhattacharjee, A.; Zhang, M.; Pourasgari, F.; Hardy, A.B.; Taylor, K.M.; et al. Characterization of Zinc Influx Transporters (ZIPs) in Pancreatic β Cells: Roles in Regulating Cytosolic Zinc Homeostasis and Insulin Secretion. J. Biol. Chem. 2015, 290, 18757–18769. [Google Scholar] [CrossRef]
- Robinson, G.L.; Cordle, S.R.; Henderson, E.; Weil, P.A.; Teitelman, G.; Stein, R. Isolation and characterization of a novel transcription factor that binds to and activates insulin control element-mediated expression. Mol. Cell Biol. 1994, 14, 6704–6714. [Google Scholar] [CrossRef]
- Robinson, G.L.; Henderson, E.; Massari, M.E.; Murre, C.; Stein, R. c-jun inhibits insulin control element-mediated transcription by affecting the transactivation potential of the E2A gene products. Mol. Cell Biol. 1995, 15, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, T.J.; Nicholson, S. Autoantibodies against human insulin. Br. Med. J. 1984, 288, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Verge, C.F.; Stenger, D.; Bonifacio, E.; Colman, P.G.; Pilcher, C.; Bingley, P.J.; Eisenbarth, G.S. Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: Combinatorial Islet Autoantibody Workshop. Diabetes 1998, 47, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. Rheumatic fever, autoimmunity, and molecular mimicry: The streptococcal connection. Int. Rev. Immunol. 2014, 33, 314–329. [Google Scholar] [CrossRef]
- Vreugdenhil, G.R.; Geluk, A.; Ottenhoff, T.H.; Melchers, W.J.; Roep, B.O.; Galama, J.M. Molecular mimicry in diabetes mellitus: The homologous domain in coxsackie B virus protein 2C and islet autoantigen GAD65 is highly conserved in the coxsackie B-like enteroviruses and binds to the diabetes associated HLA-DR3 molecule. Diabetologia 1998, 41, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Varela-Calvino, R.; Skowera, A.; Arif, S.; Peakman, M. Identification of a naturally processed cytotoxic CD8 T-cell epitope of coxsackievirus B4, presented by HLA-A2.1 and located in the PEVKEK region of the P2C nonstructural protein. J. Virol. 2004, 78, 13399–13408. [Google Scholar] [CrossRef]
- Pulli, T.; Lankinen, H.; Roivainen, M.; Hyypiä, T. Antigenic sites of coxsackievirus A9. Virology 1998, 240, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Varela-Calvino, R.; Tree, T.I.; Peakman, M. HLA Class II molecules on haplotypes associated with type 1 diabetes exhibit similar patterns of binding affinities for coxsackievirus P2C peptides. Immunology 2005, 116, 337–346. [Google Scholar] [CrossRef]
- Marttila, J.; Juhela, S.; Vaarala, O.; Hyöty, H.; Roivainen, M.; Hinkkanen, A.; Vilja, P.; Simell, O.; Ilonen, J. Responses of coxsackievirus B4-specific T-cell lines to 2C protein-characterization of epitopes with special reference to the GAD65 homology region. Virology 2001, 284, 131–141. [Google Scholar] [CrossRef]
- Marttila, J.; Hyöty, H.; Vilja, P.; Härkönen, T.; Alho, A.; Roivainen, M.; Hyypiä, T.; Ilonen, J. T cell epitopes in coxsackievirus B4 structural proteins concentrate in regions conserved between enteroviruses. Virology 2002, 293, 217–224. [Google Scholar] [CrossRef]
- Marttila, J.; Hyöty, H.; Näntö-Salonen, K.; Simell, O.; Ilonen, J. Epitopes recognized by CBV4 responding T cells: Effect of type 1 diabetes and associated HLA-DR-DQ haplotypes. Virology 2004, 319, 27–35. [Google Scholar] [CrossRef]
- Cello, J.; Strannegard, O.; Svennerholm, B. A study of the cellular immune response to enteroviruses in humans: Identification of cross-reactive T cell epitopes on the structural proteins of enteroviruses. J. Gen. Virol. 1996, 77, 2097–2108. [Google Scholar] [CrossRef]
- Simonen-Tikka, M.L.; Hiekka, A.K.; Klemola, P.; Poussa, T.; Ludvigsson, J.; Korpela, R.; Vaarala, O.; Roivainen, M. Early human enterovirus infections in healthy Swedish children participating in the PRODIA pilot study. J. Med. Virol. 2012, 84, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Nield, A.; Chew, G.S.; Myers, M.A. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PLoS ONE 2013, 8, e79316. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Moraska, A.; Cunningham, M. Alterations in major histocompatibility complex association of myocarditis induced by coxsackievirus B3 mutants selected with monoclonal antibodies to group A streptococci. Proc. Natl. Acad. Sci. USA 1994, 91, 5543–5547. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Cunningham, M.W. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell-dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsackieviral myocarditis. J. Immunol. 1996, 156, 3528–3534. [Google Scholar] [CrossRef]
- Lönnrot, M.; Hyöty, H.; Knip, M.; Roivainen, M.; Kulmala, P.; Leinikki, P.; Akerblom, H.K. Antibody cross-reactivity induced by the homologous regions in glutamic acid decarboxylase (GAD65) and 2C protein of coxsackievirus B4. Childhood Dia-betes in Finland Study Group. Clin. Exp. Immunol. 1996, 104, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Real-Fernández, F.; Gallo, A.; Nuti, F.; Altamore, L.; Del Vescovo, G.G.; Traldi, P.; Ragazzi, E.; Rovero, P.; Lapolla, A.; Papini, A.M. Cross-reactive peptide epitopes of Enterovirus Coxsackie B4 and human glutamic acid decarboxylase detecting anti-bodies in latent autoimmune diabetes in adults versus type 1 diabetes. Clin. Chim. Acta 2021, 515, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Gottumukkala, R.V.; Lv, H.; Cornivelli, L.; Wagers, A.J.; Kwong, R.Y.; Bronson, R.; Stewart, G.C.; Schulze, P.C.; Chutkow, W.; Wolpert, H.A.; et al. Myocardial infarction triggers chronic cardiac autoimmunity in type 1 diabetes. Sci. Transl. Med. 2012, 4, 138ra80. [Google Scholar] [CrossRef] [PubMed]
- Lipes, M.A.; Galderisi, A. Cardiac autoimmunity as a novel biomarker, mediator, and therapeutic target of heart disease in type 1 diabetes. Curr. Diab. Rep. 2015, 15, 30. [Google Scholar] [CrossRef]
- Eckel, R.H.; Eisenbarth, G.S. Autoimmune diabetes inflames the heart. Sci. Transl. Med. 2012, 4, 138fs18. [Google Scholar] [CrossRef] [PubMed]
- Iddings, A.C.; Shenoi, A.N.; Morales Pozzo, A.; Kiessling, S.G. Hemolytic uremic syndrome complicated by Clostridium septicum bacteremia and new-onset type 1 diabetes mellitus. Report of a case. Clin. Nephrol. 2017, 87, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Mirzai, S.; Rifai, A.O.; Webb, S.; Rifai, K.; Reiner, A. Probable Clostridium septicum pneumocephalus in a user of natural remedies with newly diagnosed diabetes mellitus type 1. IDCases 2019, 17, e00581. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xu, Y.; Ji, J.; Wei, J.; Jiang, Y.; Yang, Y.; Yang, M.; Huang, H.; Zou, R.; Fang, C.; et al. Intestinal microbiota has important effect on severity of hand foot and mouth disease in children. BMC Infect. Dis. 2021, 21, 1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Mou, D.; Li, T.; Chen, Z.; Ma, C.; Liang, L.; He, Q. Integrated analysis reveals important differences in the gut and oropharyngeal microbiota between children with mild and severe hand, foot, and mouth disease. Emerg. Microbes Infect. 2023, 12, 2192819. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Lan, Z.; Wen, Y.; Zheng, C.; Rong, Z.; Liu, T.; Chen, S.; Yang, X.; Zheng, H.; Wu, W. Synbiotics Supplements Lower the Risk of Hand, Foot, and Mouth Disease in Children, Potentially by Providing Resistance to Gut Microbiota Dysbiosis. Front. Cell Infect. Microbiol. 2021, 11, 729756. [Google Scholar] [CrossRef]
- Muniain-Mujika, I.; Calvo, M.; Lucena, F.; Girones, R. Comparative analysis of viral pathogens and potential indicators in shellfish. Int. J. Food Microbiol. 2003, 83, 75–85. [Google Scholar] [CrossRef] [PubMed]
- LaBelle, R.L.; Gerba, C.P.; Goyal, S.M.; Melnick, J.L.; Cech, I.; Bogdan, G.F. Relationships between environmental factors, bacterial indicators, and the occurrence of enteric viruses in estuarine sediments. Appl. Environ. Microbiol. 1980, 39, 588–596. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Podufaly, A. Autoreactive T-cell receptor (Vbeta/D/Jbeta) sequences in diabetes recognize insulin, the insulin receptor, and each other, and are targets of insulin antibodies. Open Autoimmun. J. 2012, 4, 10–22. [Google Scholar] [CrossRef]
- Rudy, G.; Stone, N.; Harrison, L.C.; Colman, P.G.; McNair, P.; Brusic, V.; French, M.B.; Honeyman, M.C.; Tait, B.; Lew, A.M. Similar peptides from two beta cell autoantigens, proinsulin and glutamic acid decarboxylase, stimulate T cells of individuals at risk for insulin-dependent diabetes. Mol. Med. 1995, 1, 625–633. [Google Scholar] [CrossRef]
- Geluk, A.; van Meijgaarden, K.E.; Schloot, N.C.; Drijfhout, J.W.; Ottenhoff, T.H.; Roep, B.O. HLA-DR binding analysis of peptides from islet antigens in IDDM. Diabetes 1998, 47, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Schloot, N.C.; Roep, B.O.; Wegmann, D.R.; Yu, L.; Wang, T.B.; Eisenbarth, G.S. T-cell reactivity to GAD65 peptide sequences shared with coxsackie virus protein in recent-onset IDDM, post-onset IDDM patients and control subjects. Diabetologia 1997, 40, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Tay, L.; Yin-Murphy, M.; Chua, P.H.; Koh, L.H. Prevalence of coxsackievirus B antibody in patients with suspected rheumatic fever and rheumatic heart disease. Singap. Med. J. 1983, 24, 37–40. [Google Scholar]
- Pongpanich, B.; Boonpucknavig, S.; Wasi, C.; Tanphaichitr, P.; Boonpucknavig, V. Immunopathology of acute rheumatic fever and rheumatic heart disease. The demonstration of Coxsackie group B viral antigen in the myocardium. Clin. Rheumatol. 1983, 2, 217–222. [Google Scholar] [CrossRef]
- Kaznacheev, V.P.; Iavorovskaia, V.E. Bacterial-viral coalition in the etiology of rheumatic heart disease. Ter. Arkh. 1973, 45, 15–23. (In Russian) [Google Scholar] [PubMed]
- Cunningham, M.W. T cell mimicry in inflammatory heart disease. Mol. Immunol. 2004, 40, 1121–1127. [Google Scholar] [CrossRef]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular mimicry in virus infection: Crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; Oldstone, M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef]
- Davies, J.M. Molecular mimicry: Can. epitope mimicry induce autoimmune disease? Immunol. Cell Biol. 1997, 75, 113–126. [Google Scholar] [CrossRef]
- Rose, N.R.; Mackay, I.R. Molecular mimicry: A critical look at exemplary instances in human diseases. Cell Mol. Life Sci. 2000, 57, 542–551. [Google Scholar] [CrossRef]
- Benoist, C.; Mathis, D. Autoimmunity provoked by infection: How good is the case for T cell epitope mimicry? Nat. Immunol. 2001, 2, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Fourneau, J.M.; Bach, J.M.; van Endert, P.M.; Bach, J.F. The elusive case for a role of mimicry in autoimmune diseases. Mol. Immunol. 2004, 40, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Damian, R.T. Molecular Mimicry in Biological Adaptation. Science 1965, 147, 824. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.; Tsunoda, I.; Fujinami, R.S. Multiple sclerosis and virus induced immune responses: Autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 2006, 39, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Chang, C.; Gershwin, M.E.; Anaya, J.M. Bystander activation and autoimmunity. J. Autoimmun. 2019, 103, 102301. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R. Negative selection, epitope mimicry and autoimmunity. Curr. Opin. Immunol. 2017, 49, 51–55. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Synergistic Activation of Toll-Like and NOD Receptors by Complementary Antigens as Facilitators of Autoimmune Disease: Review, Model and Novel Predictions. Int. J. Mol. Sci. 2020, 21, 4645. [Google Scholar] [CrossRef]
- Root-Bernstein, R. Innate Receptor Activation Patterns Involving TLR and NLR Synergisms in COVID-19, ALI/ARDS and Sepsis Cytokine Storms: A Review and Model Making Novel Predictions and Therapeutic Suggestions. Int. J. Mol. Sci. 2021, 22, 2108. [Google Scholar] [CrossRef]
- Root-Bernstein, R. From Co-Infections to Autoimmune Disease via Hyperactivated Innate Immunity: COVID-19 Autoimmune Coagulopathies, Autoimmune Myocarditis and Multisystem Inflammatory Syndrome in Children. Int. J. Mol. Sci. 2023, 24, 3001. [Google Scholar] [CrossRef]
- Plotz, P.H. Autoantibodies are anti-idiotype antibodies to antiviral antibodies. Lancet 1983, 2, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Tzioufas, A.G.; Routsias, J.G. Idiotype, anti-idiotype network of autoantibodies: Pathogenetic considerations and clinical application. Autoimmun. Rev. 2010, 9, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Westall, F.C.; Root-Bernstein, R.S. An explanation of prevention and suppression of experimental allergic encephalomyelitis. Mol. Immunol. 1983, 20, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Westall, F.C.; Root-Bernstein, R. Cause and prevention of postinfectious and postvaccinal neuropathies in light of a new theory of autoimmunity. Lancet 1986, 2, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Multiple-antigen-mediated autoimmunity (MAMA) in AIDS: A possible model for postinfectious autoimmune complications. Res. Immunol. 1990, 141, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.W.; Kion, T.A.; Grant, M.D. An idiotypic network model of AIDS immunopathogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 3060–3064. [Google Scholar] [CrossRef] [PubMed]
- Süsal, C.; Hoffman, G.W.; Daniel, V.; Grant, M.; Opelz, G. Complementarities and network interactions in AIDS. J. Autoimmun. 1993, 6, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Pendergraft, W.F., 3rd; Preston, G.A.; Shah, R.R.; Tropsha, A.; Carter, C.W., Jr.; Jennette, J.C.; Falk, R.J. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004, 10, 72–79. [Google Scholar] [CrossRef]
- Pendergraft, W.F., 3rd; Pressler, B.M.; Jennette, J.C.; Falk, R.J.; Preston, G.A. Autoantigen complementarity: A new theory implicating complementary proteins as initiators of autoimmune disease. J. Mol. Med. 2005, 83, 12–25. [Google Scholar] [CrossRef]
- Yang, J.; Bautz, D.J.; Lionaki, S.; Hogan, S.L.; Chin, H.; Tisch, R.M.; Schmitz, J.L.; Pressler, B.M.; Jennette, J.C.; Falk, R.J.; et al. ANCA patients have T cells responsive to complementary PR-3 antigen. Kidney Int. 2008, 74, 1159–1169. [Google Scholar] [CrossRef]
- Reynolds, J.; Preston, G.A.; Pressler, B.M.; Hewins, P.; Brown, M.; Roth, A.; Alderman, E.; Bunch, D.; Jennette, J.C.; Cook, H.T.; et al. Autoimmunity to the alpha 3 chain of type IV collagen in glomerulonephritis is triggered by ‘autoantigen complementarity’. J. Autoimmun. 2015, 59, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R. How to Make a Non-Antigenic Protein (Auto) Antigenic: Molecular Complementarity Alters Antigen Processing and Activates Adaptive-Innate Immunity Synergy. Anticancer Agents Med. Chem. 2015, 15, 1242–1259. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Krupovic, M.; Forterre, P. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. J. Bacteriol. 2018, 200, e00580-17. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, R.M.; Schluter, S.F.; Bernstein, H.; Marchalonis, J.J. Primordial emergence of the recombination activating gene 1 (RAG1): Sequence of the complete shark gene indicates homology to microbial integrases. Proc. Natl. Acad. Sci. USA 1996, 93, 9454–9459. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Eastman, Q.M.; Schatz, D.G. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature 1998, 394, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Pontarotti, P.; Nehari, S.; Raoult, D. ‘Cannibalism’ of exogenous DNA sequences: The ancestral form of adaptive immunity which entails recognition of danger. Front. Immunol. 2022, 13, 989707. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.; Dray, S. Conversion of non-immune rabbit spleen cells by ribonucleic acid of lymphoid cells from an immunized rabbit to produce IgM and IgG antibody of foreign heavy-chain allotype. J. Immunol. 1971, 107, 83–95. [Google Scholar] [CrossRef]
- Adler, F.L.; Fishman, M.; Dray, S. Antibody formation initiated in vitro. 3. Antibody formation and allotypic specificity directed by ribonucleic acid from peritoneal exudate cells. J. Immunol. 1966, 97, 554–558. [Google Scholar] [CrossRef]
- Fainboim, L.; Sztein, M.B.; Serrate, S.; Satz, L. In vitro transfer of cellular immunity in experimental allergic orchitis by means of immune RNA. Immunology 1979, 38, 311–316. [Google Scholar]
- Satz, M.L.; Sztein, M.B.; Serrate, S.; Braun, M. Mechanism of immune transfer by RNA ex-tracts. Immune RNA induces the synthesis of idiotype-bearing antigen receptors in non-committed cells. Mol. Cell Biochem. 1980, 33, 105–113. [Google Scholar] [CrossRef]
- Theurer, K. Eine neue Instruktionstheorie. Möglichkeiten einer rückläufigen infor-mationsübertragung von Polypeptidsequenzen auf RNA, insbesondere bei der Anti-körpersynthese, sowie Mechanismen der Toleranzerzeugung und Immunsuppression [A new instruction theory: Possibility of a reverse flow of information from polypeptide se-quences to RNA particularly in antibody synthesis, and the mechanisms of tolerance in-duction and immunosuppression (author’s transl)]. Infection 1975, 3, 178–182. (In German) [Google Scholar] [CrossRef]
- Root Bernstein, R.S. Self, Nonself, and the Paradoxes of Autoimmunity. In Organism and the Development of Self; Tauber, A.I., Ed.; Kluwer: Boston, MA, USA, 1991; pp. 159–209. [Google Scholar]
- Viza, D.; Fudenberg, H.H.; Palareti, A.; Ablashi, D.; De Vinci, C.; Pizza, G. Transfer factor: An overlooked potential for the prevention and treatment of infectious diseases. Folia Biol. 2013, 59, 53–67. [Google Scholar]
- Root-Bernstein, R. The Mysterious, and Potentially Revolutionary, Immunological Properties of Transfer Factor: A Review. Preprints. 2019, p. 2019050386. Available online: https://www.preprints.org/manuscript/201905.0386/v1 (accessed on 24 January 2024).
- Hara, N.; Alkanani, A.K.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Prevention of virus-induced type 1 diabetes with antibiotic therapy. J. Immunol. 2012, 189, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Viza, D.; Boucheix, C.; Kern, D.H.; Pilch, Y.H. Human lymphoblastoid cells in culture replicate immune information carried by xenogeneic RNA. Differentiation 1978, 11, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Passos Júnior, G.A.; de Lucca, F.L. RNA-mediated transfer of cellular immunity to a synthetic env antigen of the human immunodeficiency virus (HIV-1). Mol. Cell Biochem. 1991, 108, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Fraumene, C.; Manghina, V.; Silverman, M.; Uzzau, S. Clostridial Butyrate Biosynthesis Enzymes Are Significantly Depleted in the Gut Microbiota of Nonobese Diabetic Mice. mSphere 2018, 3, e00492-18. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Shan, K.; Pan, L.L.; Feng, N.; Lv, Z.; Sun, Y.; Li, J.; Wu, C.; Zhang, H.; Chen, W.; et al. Clostridium butyricum CGMCC0313.1 Protects against Autoimmune Diabetes by Modu-lating Intestinal Immune Homeostasis and Inducing Pan-creatic Regulatory T Cells. Front. Immunol. 2017, 8, 1345. [Google Scholar] [CrossRef]
- Jia, L.; Li, D.; Feng, N.; Shamoon, M.; Sun, Z.; Ding, L.; Zhang, H.; Chen, W.; Sun, J.; Chen, Y.Q. Anti-diabetic Effects of Clos-tridium butyricum CGMCC0313.1 through Promoting the Growth of Gut Butyrate-producing Bacteria in Type 2 Diabetic Mice. Sci. Rep. 2017, 7, 7046. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.Y.; Liu, Y. Effect of fecal microbiota transplantation on type 1 diabetes mellitus in non-obese diabetic mice and its underlying mechanism. Zhonghua Yi Xue Za Zhi 2022, 102, 1224–1231. (In Chinese) [Google Scholar] [CrossRef]
- Rafeek, R.A.M.; Sikder, S.; Hamlin, A.S.; Andronicos, N.M.; McMillan, D.J.; Sriprakash, K.S.; Ketheesan, N. Requirements for a Robust Animal Model to Investigate the Disease Mechanism of Autoimmune Complications Associated With ARF/RHD. Front. Cardiovasc. Med. 2021, 8, 675339. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S.; Rose, N.R. Viruses as adjuvants for autoimmunity: Evidence from Coxsackievirus-induced myocarditis. Rev. Med. Virol. 2005, 15, 17–27. [Google Scholar] [CrossRef]
- Cunningham, M.W. Molecular Mimicry, Autoimmunity, and Infection: The Cross-Reactive Antigens of Group A Streptococci and their Sequelae. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Huber, J.; Ziehl, A.; Pietrowicz, M. SARS-CoV-2 and Its Bacterial Co- or Super-Infections Synergize to Trigger COVID-19 Autoimmune Cardiopathies. Int. J. Mol. Sci. 2023, 24, 12177. [Google Scholar] [CrossRef] [PubMed]
- Allakany, A.I.; Elbanna, A.A.; Rohoma, K.H.; Ahmed, S.M.; Ibrahim, A.E.; Fawzy, M.A.; Header, D.A. Study of the gut microbiome in Egyptian patients with type 1 diabetes mellitus. Prz. Gastroenterol. 2023, 18, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Tamahane, V.; Bhanushali, S.; Shah, N.; Gupta, A.; Khadilkar, V.; Gondhalekar, K.; Khadil-kar, A.; Shouche, Y. A comparative study of the gut microbiome in Indian children with type 1 diabetes and healthy controls. J. Diabetes 2023. [Google Scholar] [CrossRef] [PubMed]
- Gil-Cruz, C.; Perez-Shibayama, C.; De Martin, A.; Ronchi, F.; van der Borght, K.; Niederer, R.; Onder, L.; Lütge, M.; Novkovic, M.; Nindl, V.; et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 2019, 366, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.F.; Zhang, W.Y.; Wen, Q.; Chen, W.J.; Wang, Z.M.; Chen, J.; Zhu, F.; Liu, K.; Cheng, L.X.; Yang, J.; et al. Fecal microbiota transplantation alleviates myocardial damage in myocarditis by restoring the microbiota composition. Pharmacol. Res. 2019, 139, 412–421. [Google Scholar] [CrossRef]
- Neu, U.; Mainou, B.A. Virus interactions with bacteria: Partners in the infectious dance. PLoS Pathog. 2020, 16, e1008234. [Google Scholar] [CrossRef]
- Usviatsov, B.; Pan’kov, A.S.; Bukharin, O.V. Mechanisms of interaction of associative symbionts during viral-bacterial infections. Zh Mikrobiol Epidemiol Immunobiol. 2009, 2, 117–121. [Google Scholar]
- Aguilera, E.R.; Nguyen, Y.; Sasaki, J.; Pfeiffer, J.K. Bacterial Stabilization of a Panel of Picornaviruses. mSphere 2019, 4, e00183-19. [Google Scholar] [CrossRef]
- Waldman, P.; Meseguer, A.; Lucas, F.; Moulin, L.; Wurtzer, S. Interaction of Human Enteric Viruses with Microbial Compounds: Implication for Virus Persistence and Disinfection Treatments. Environ. Sci. Technol. 2017, 51, 13633–13640. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ma, W.-T.; Pang, M.; Fan, Q.-L.; Hua, J.-L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Xiong, D.; Shi, J.; Long, M.; Chen, Z. The Interaction Between Viruses and Intestinal Microbiota: A Review. Curr. Microbiol. 2021, 78, 3597–3608. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M. Enteric viruses exploit the microbiota to promote infection. Curr. Opin. Virol. 2019, 37, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Barin, J.G.; Talor, M.V.; Diny, N.L.; Ong, S.; Schaub, J.A.; Gebremariam, E.; Bedja, D.; Chen, G.; Choi, H.S.; Hou, X.; et al. Regulation of autoimmune myocarditis by host responses to the microbiome. Exp. Mol. Pathol. 2017, 103, 141–152. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Chen, R.; Zhang, B.; Zhang, S.; Khan, B.A.; Zhu, D.; Wu, Z.; Xiao, C.; Chen, B.; Chen, F.; et al. Fecal microbiota transplantation treatment of autoimmune-mediated type 1 diabetes mellitus. Front. Immunol. 2022, 13, 930872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Deng, F.; Chen, J.; Chen, F.; Wu, Z.; Li, L.; Hou, K. Fecal microbiota transplantation treatment of autoimmune-mediated type 1 diabetes: A systematic review. Front. Cell Infect. Microbiol. 2022, 12, 1075201. [Google Scholar] [CrossRef]
- Hadj Hassine, I.; Gharbi, J.; Amara, I.; Alyami, A.; Subei, R.; Almalki, M.; Hober, D.; M’hadheb, M.B. Cloning and Molecular Characterization of the Recombinant CVB4E2 Immunogenic Viral Protein (rVP1), as a Potential Subunit Protein for Vaccine and Immunodiagnostic Reagent Candidate. Microorganisms 2023, 11, 1192. [Google Scholar] [CrossRef]
- Gharbi, J.; Almalki, M.A.; Ben M’hadheb, M. The introduction of mutations in the wild type coxsackievirus B3 (CVB3) IRES RNA leads to different levels of in vitro reduced replicative and translation efficiencies. PLoS ONE 2022, 17, e0274162. [Google Scholar] [CrossRef]
- Hankaniemi, M.M.; Stone, V.M.; Andrejeff, T.; Heinimäki, S.; Sioofy-Khojine, A.B.; Marjomäki, V.; Hyöty, H.; Blazevic, V.; Flodström-Tullberg, M.; Hytönen, V.P.; et al. Formalin treatment increases the stability and immunogenicity of coxsackievirus B1 VLP vaccine. Antivir. Res. 2019, 171, 104595. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, C.; Li, J.; Dai, Z.; Huang, J.; Deng, F.; Wang, X.; Yue, X.; Hu, X.; Li, Y.; et al. Designing a multi-epitope vaccine against coxsackievirus B based on immunoinformatics approaches. Front. Immunol. 2022, 13, 933594. [Google Scholar] [CrossRef]
- Gharbi, J.; Hadj Hassine, I.; Hassine, M.; Al-Malki, M.; Al-Yami, A.; Al-Bachir, A.; Ben M’hadheb, M. Viral Protein VP1 Virus-like Particles (VLP) of CVB4 Induces Protective Immunity against Lethal Challenges with Diabetogenic E2 and Wild Type JBV Strains in Mice Model. Viruses 2023, 15, 878. [Google Scholar] [CrossRef] [PubMed]
- Hyöty, H.; Leon, F.; Knip, M. Developing a vaccine for type 1 diabetes by targeting coxsackievirus B. Expert. Rev. Vaccines 2018, 17, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Drescher, K.M.; von Herrath, M.; Tracy, S. Enteroviruses, hygiene and type 1 diabetes: Toward a preventive vaccine. Rev. Med. Virol. 2015, 25, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.G.; Lakshmikanth, T.; Laitinen, O.H.; Utorova, R.; Jacobson, S.; Oikarinen, M.; Domsgen, E.; Koivunen, M.R.; Chaux, P.; Devard, N.; et al. A preclinical study on the efficacy and safety of a new vaccine against Coxsackievirus B1 reveals no risk for accelerated diabetes development in mouse models. Diabetologia 2015, 58, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Stone, V.M.; Butrym, M.; Hankaniemi, M.M.; Sioofy-Khojine, A.B.; Hytönen, V.P.; Hyöty, H.; Flodström-Tullberg, M. Coxsackievirus B Vaccines Prevent Infection-Accelerated Diabetes in NOD Mice and Have No Disease-Inducing Effect. Diabetes 2021, 70, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Höfling, K.; Kim, K.S.; Leser, J.S.; Chapman, N.M.; Willian, S.; Gauntt, C.J.; Tracy, S. Progress toward vaccines against viruses that cause heart disease. Herz 2000, 25, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Tracy, S. Can recombinant DNA technology provide useful vaccines against viruses which induce heart disease? Eur. Heart J. 1995, 16, 144–146. [Google Scholar] [CrossRef]
- Zhang, L.; Parham, N.J.; Zhang, F.; Aasa-Chapman, M.; Gould, E.A.; Zhang, H. Vaccination with coxsackievirus B3 virus-like particles elicits humoral immune response and protects mice against myocarditis. Vaccine 2012, 30, 2301–2308. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Li, H.; Liu, L. Hand-Foot-and-Mouth Disease-Associated Enterovirus and the Development of Multivalent HFMD Vaccines. Int. J. Mol. Sci. 2022, 24, 169. [Google Scholar] [CrossRef]
- Henke, A.; Jarasch, N.; Martin, U.; Wegert, J.; Wildner, A.; Zell, R.; Wutzler, P. Recombinant coxsackievirus vectors for prevention and therapy of virus-induced heart disease. Int. J. Med. Microbiol. 2008, 298, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.V.; Lyras, D.; Douce, G.R. Status of vaccine research and development for Clostridium difficile. Vaccine 2019, 37, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, J.; Zorman, J.; Wang, S.; Miezeiewski, M.; Xie, J.; Soring, K.; Petrescu, I.; Rogers, I.; Thiriot, D.S.; Cook, J.C.; et al. Development of a recombinant toxin fragment vaccine for Clostridium difficile infection. Vaccine 2014, 32, 2812–2818. [Google Scholar] [CrossRef] [PubMed]
- Foglia, G.; Shah, S.; Luxemburger, C.; Pietrobon, P.J. Clostridium difficile: Development of a novel candidate vaccine. Vaccine 2012, 30, 4307–4309. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.H.; Glenn, G.; Flyer, D.; Zhou, B.; Liu, Y.; Sullivan, E.; Wu, H.; Cummings, J.F.; Elllingsworth, L.; Smith, G. Clostridium difficile chimeric toxin receptor binding domain vaccine induced protection against different strains in active and passive challenge models. Vaccine 2017, 35, 4079–4087. [Google Scholar] [CrossRef]
- Anosova, N.G.; Brown, A.M.; Li, L.; Liu, N.; Cole, L.E.; Zhang, J.; Mehta, H.; Kleanthous, H. Systemic antibody responses induced by a two-component Clostridium difficile toxoid vaccine protect against C. difficile-associated disease in hamsters. J. Med. Microbiol. 2013, 62, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Heuler, J.; Chandra, H.; Sun, X. Mucosal Vaccination Strategies against Clostridioides difficile Infection. Vaccines 2023, 11, 887. [Google Scholar] [CrossRef] [PubMed]
- Steer, A.C.; Carapetis, J.R.; Dale, J.B.; Fraser, J.D.; Good, M.F.; Guilherme, L.; Moreland, N.J.; Mulholland, E.K.; Schodel, F.; Smeesters, P.R. Status of research and development of vaccines for Streptococcus pyogenes. Vaccine 2016, 34, 2953–2958. [Google Scholar] [CrossRef]
- Pandey, M.; Good, M.F. The quest for GAS vaccine. Oncotarget 2015, 6, 34063–34064. [Google Scholar] [CrossRef]
- Giffard, P.M.; Tong, S.Y.C.; Holt, D.C.; Ralph, A.P.; Currie, B.J. Concerns for efficacy of a 30-valent M-protein-based Streptococcus pyogenes vaccine in regions with high rates of rheumatic heart disease. PLoS Negl. Trop. Dis. 2019, 13, e0007511. [Google Scholar] [CrossRef]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [PubMed]
- Probert, C.S.; Chott, A.; Turner, J.R.; Saubermann, L.J.; Stevens, A.C.; Bodinaku, K.; Elson, C.O.; Balk, S.P.; Blumberg, R.S. Persistent clonal expansions of peripheral blood CD4+ lymphocytes in chronic inflammatory bowel disease. J. Immunol. 1996, 157, 3183–3191. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Maciejewski, J.P.; Chen, G.; Young, N.S. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J. Clin. Investig. 2001, 108, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, I.Z.; Britanova, O.V.; Bolotin, D.A.; Chkalina, A.V.; Staroverov, D.B.; Zvyagin, I.V.; Kotlobay, A.A.; Turchaninova, M.A.; Fedorenko, D.A.; Novik, A.A.; et al. Quantitative tracking of T cell clones after haematopoietic stem cell transplantation. EMBO Mol. Med. 2011, 3, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.D.; Klein, J.T.; Silver, R.F.; Liu, M.C.; Greenlee, B.M.; Moller, D.R. Selective activation and accumulation of oligoclonal V beta-specific T cells in active pulmonary sarcoidosis. J. Clin. Investig. 1994, 94, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.; Vignali, M.; Klinger, M.; Dines, J.; Kaplan, I.; Svejnoha, E.; Craft, T.; Boland, K.; Pesesky, M.; Gittelman, R.M.; et al. A Large-Scale Database of T-Cell Receptor Beta (TCRb) Sequences and Binding Associations from Natural and Synthetic Exposure to SARS-CoV-2. 2020. Available online: https://clients.adaptivebiotech.com/pub/covid-2020 (accessed on 20 November 2022).
- Durinovic-Bello, I.; Steinle, A.; Ziegler, A.G.; Schendel, D.J. 1994. HLA-DQ-restricted, islet-specific T-cell clones of a type I diabetic patient. T-cell receptor sequence similarities to insulitis-inducing T-cells of nonobese diabetic mice. Diabetes 1994, 43, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Kent, S.C.; Chen, Y.; Bregoli, L.; Clemmings, S.M.; Kenyon, N.S.; Ricordi, C.; Hering, B.J.; Hafler, D.A. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 2005, 435, 224–228. [Google Scholar] [CrossRef]
- Seay, H.R.; Yusko, E.; Rothweiler, S.J.; Zhang, L.; Posgai, A.L.; Campbell-Thompson, M.; Vignali, M.; Emerson, R.O.; Kaddis, J.S.; Ko, D.; et al. Tissue distribution and clonal diversity of the T and B cell repertoire in type 1 diabetes. JCI Insight 2016, 1, e88242. [Google Scholar] [CrossRef]
- Luppi, P.; Rudert, W.; Licata, A.; Riboni, S.; Betters, D.; Cotrufo, M.; Frati, G.; Condorelli, G.; Trucco, M. Expansion of specific alphabeta+ T-cell subsets in the myocardium of patients with myocarditis and idiopathic dilated cardiomyopathy associated with Coxsackievirus B infection. Hum. Immunol. 2003, 64, 194–210. [Google Scholar] [CrossRef]

















| TCR | Cox A | Cox B | Clost | InsRec | Ins | GlucRec | Gluc | PTPRN | GAD | Other Diab | Myosin | Actin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | X | X | X | X | ||||||||
| A2 | X | X | X | X | ||||||||
| A3 | X | X | X | X | ||||||||
| A4 | X | X | ||||||||||
| A5 | X | X | X | |||||||||
| A6 | X | X | X | X | X | X | ||||||
| A7 | X | X | X | |||||||||
| A8 | X | X | X | X | X | |||||||
| A9 | X | X | X | |||||||||
| A10 | X | X | X | X | ||||||||
| A11 | X | X | X | |||||||||
| DIA 1 | X | X | X | X | X | X | ||||||
| DIA 2 | X | X | X | X | X | |||||||
| DIA 3 | X | X | X | X | X | X | ||||||
| DIA 4 | X | X | ||||||||||
| DIA 5 | X | X | X | X | ||||||||
| DIA 6 | X | X | X | X | ||||||||
| DIA 7 | X | X | X | X | ||||||||
| DIA 8 | X | X | X | X | X | X | ||||||
| DIA 9 | X | X | X | |||||||||
| DIA 10 | X | X | X | X | X | |||||||
| DIA 11 | X | X | X | X | X | |||||||
| K2.4 | X | X | X | X | X | X | ||||||
| K2.12 | X | X | X | X | ||||||||
| K2.16 | X | X | X | X |
| TCR | Cox A | Cox B | GAS | GBS | Strep Virid | Myo | Act | Coll | Lam | Dyn | Tit | Ryan Rec | ACh Rec | Glut Rec | Adr Rec | Ins | Glu | Other Cardiac |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2.1 | X | X | X | X | X | X | X | |||||||||||
| 2.2 | X | X | X | X | X | X | ||||||||||||
| 2.3 | X | X | X | |||||||||||||||
| 2.4 | X | X | X | X | (X) | X | X | |||||||||||
| 2.5 | X | X | X | X | (X) | X | ||||||||||||
| 2.6 | X | X | X | X | X | X | ||||||||||||
| 2.7 | X | X | X | (X) | ||||||||||||||
| 2.8 | X | X | X | X | X | |||||||||||||
| 2.9 | X | X | X | (X) | ||||||||||||||
| 2.10 | X | X | X | (X) | X | X | ||||||||||||
| 2.11 | X | X | X | (X) | ||||||||||||||
| 2.12 | X | X | X | (X) | X | |||||||||||||
| 2.13 | X | X | (X) | (X) | X | |||||||||||||
| 2.14 | X | X | ||||||||||||||||
| 2.15 | X | X | ||||||||||||||||
| 2.16 | X | X | X | (X) | X | |||||||||||||
| 2.17 | X | (X) | ||||||||||||||||
| 2.18 | X | X | (X) | X | ||||||||||||||
| 2.19 | X | X | X | X | X | |||||||||||||
| 7.1 | X | X | X | X | ||||||||||||||
| 7.2 | X | X | X | X | X | X | ||||||||||||
| 7.3 | X | X | X | X | X | |||||||||||||
| 7.4 | X | X | X | X | X | (X) | X | X | ||||||||||
| 7.5 | X | X | X | X | X | |||||||||||||
| 7.6 | X | X | X | |||||||||||||||
| 7.7 | X | X | X | X | (X) | |||||||||||||
| 7.8 | X | X | X | X | X | |||||||||||||
| 7.9 | X | X | ||||||||||||||||
| 7.10 | X | X | X | |||||||||||||||
| 7.11 | X | X | X | |||||||||||||||
| 7.12 | X | X | X | |||||||||||||||
| 7.13 | X | X | X | X | X | |||||||||||||
| 7.14 | X | X | X | |||||||||||||||
| 7.15 | X | X | X | X | X | X | X | |||||||||||
| 7.16 | X | X | X |
| Binding Const. (µM) | TCR 1 | TCR2, K2.16 | TCR4 K2.4 | TCR 8 | TCR 9 | TCR 10 | TCR4,8,9 | INS | GLUC | InsRec 105–118 | InsRec 897–915 | INS Ab | GLUC Ab | InsRec α Ab |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K2.12 | 90 | 130 | 250 | 110 | 110 | 110 | 180 | 75 | 120 | 120 | >1000 | 220 | 110 | 24 |
| 1 | 110 | 310 | 70 | 70 | 150 | 33 | 80 | >10,000 | 125 | 300 | 41 | 63 | 28 | |
| 2, K2.16 | 230 | 220 | 330 | 120 | >10,000 | >1000 | >10,000 | >1000 | >1000 | 55 | 99 | 30 | ||
| 4, K2.4 | 400 | 470 | 110 | >1000 | 15 | >1000 | >1000 | >1000 | 550 | >1000 | 3.8 | |||
| 8 | 400 | 270 | 200 | 130 | 140 | 120 | 130 | >1000 | >1000 | 88 | ||||
| 9 | >1000 | >1000 | 23 | 140 | 150 | 145 | >1000 | 2.2 | >1000 | |||||
| 10 | 90 | 130 | 90 | 110 | 140 | 33 | >1000 | >1000 | ||||||
| 4,8,9 | >1000 | >1000 | >1000 | >1000 | 12 | 5.0 | 300 |
| Kd | HS CXB4 | MN CXB3 | CXB3 MAB948 | Myosin | Laminin | Actin | Collagen IV | Vitronectin |
|---|---|---|---|---|---|---|---|---|
| GAS MA1-10698 | 4 × 10−11 | 5 × 10−9 | >10−5 | 2 × 10−10 | 2 × 10−9 | 2 × 10−6 | 1 × 10−7 | 3 × 10−8 |
| GAS MA1-10699 | 6 × 10−11 | 6 × 10−9 | >10−5 | >10−5 | >10−5 | >10−5 | >10−5 | >10−5 |
| GAS MA1-10700 | 8 × 10−11 | 4 × 10−9 | >10−5 | 3 × 10−10 | 8 × 10−6 | >10−5 | 3 × 10−7 | >10−5 |
| GAS MA1-10701 | 3 × 10−11 | 5 × 10−9 | >10−5 | >10−5 | >10−5 | >10−5 | >10−5 | >10−5 |
| GAS MBS190189 | 5 × 10−10 | 1 × 10−7 | >10−5 | 3 × 10−8 | 4 × 10−9 | >10−5 | >10−5 | >10−5 |
| CXB4 Horse | 3 × 10−8 | 3 × 10−7 | 2 × 10−9 | 2 × 10−11 | >10−5 | |||
| CXB3 Monkey | 3 × 10−8 | >10−5 | 7 × 10−10 | 3 × 10−10 | >10−5 | |||
| CXB3 MAB948 | 5 × 10−8 | 1 × 10−7 | >10−5 | >10−5 | >10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R. T-Cell Receptor Sequences Identify Combined Coxsackievirus–Streptococci Infections as Triggers for Autoimmune Myocarditis and Coxsackievirus–Clostridia Infections for Type 1 Diabetes. Int. J. Mol. Sci. 2024, 25, 1797. https://doi.org/10.3390/ijms25031797
Root-Bernstein R. T-Cell Receptor Sequences Identify Combined Coxsackievirus–Streptococci Infections as Triggers for Autoimmune Myocarditis and Coxsackievirus–Clostridia Infections for Type 1 Diabetes. International Journal of Molecular Sciences. 2024; 25(3):1797. https://doi.org/10.3390/ijms25031797
Chicago/Turabian StyleRoot-Bernstein, Robert. 2024. "T-Cell Receptor Sequences Identify Combined Coxsackievirus–Streptococci Infections as Triggers for Autoimmune Myocarditis and Coxsackievirus–Clostridia Infections for Type 1 Diabetes" International Journal of Molecular Sciences 25, no. 3: 1797. https://doi.org/10.3390/ijms25031797