
Identification of Tomato microRNAs in Late Response to Trichoderma atroviride

Abstract
:1. Introduction
2. Results
2.1. Identification and Categorization of miRNAs in Tomato Plants Treated with Trichoderma
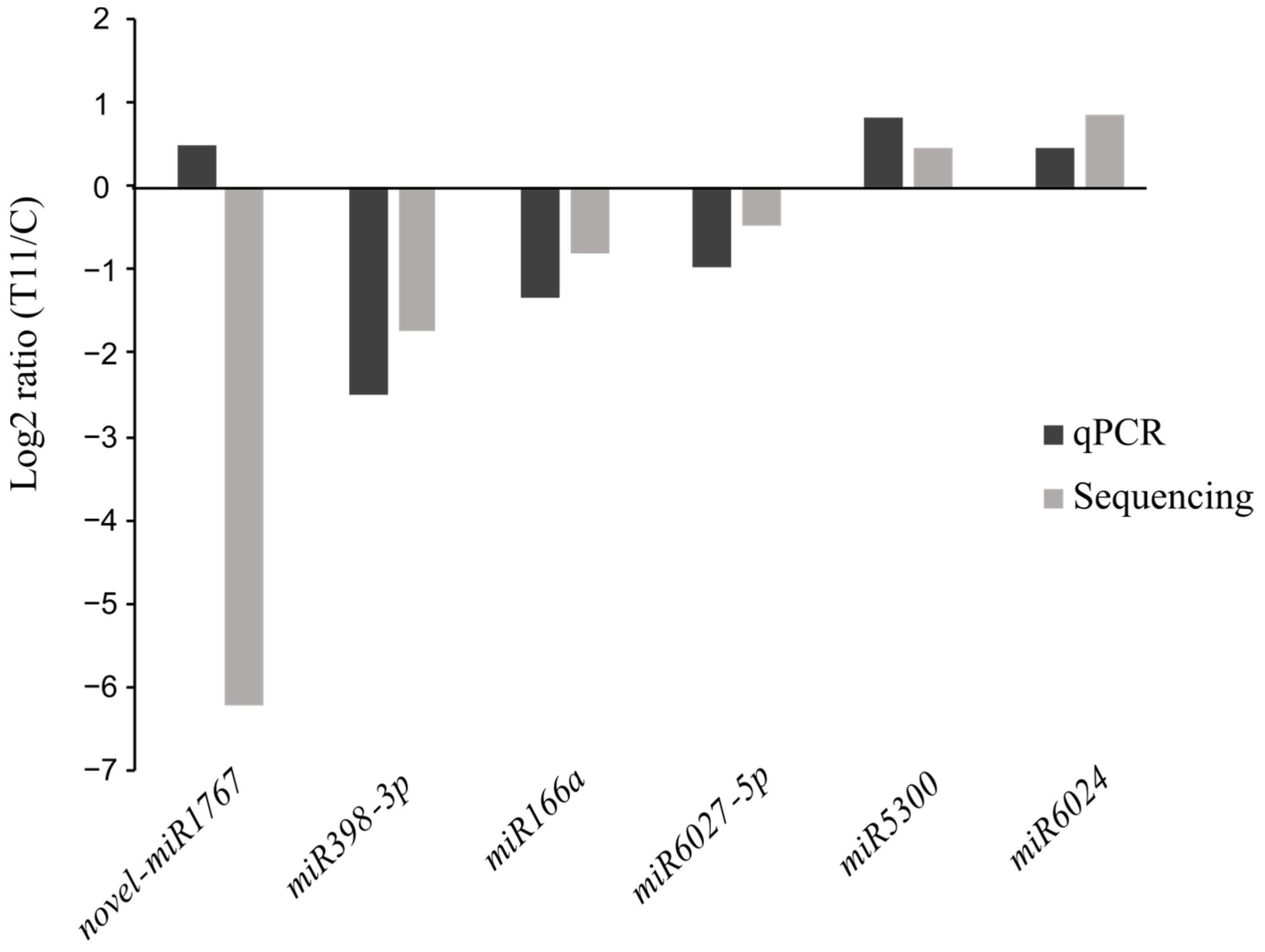
2.2. Differential Expression of miRNAs in T11-Treated Tomato Plants
2.3. Prediction of miRNA Target Genes and Their Biological Function
3. Discussion
4. Materials and Methods
4.1. Tomato Plants, T11 Inoculation, and Sample Collection
4.2. RNA Extraction and Sequencing
4.3. Data Quality Control and Identification of Known and Novel miRNA Candidates
4.4. Prediction of miRNA Target Genes and KEGG Function Analysis
4.5. Statistical Analysis of Sequencing Data and Data Visualization
4.6. Validation of miRNAs and Their Target Genes by qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woo, S.L.; Hermosa, R.; Lorito, M.; Monte, E. Trichoderma: A multipurpose, plant-beneficial microorganism for eco-sustainable agriculture. Nat. Rev. Microbiol. 2023, 21, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Morán-Diez, M.E.; Martínez de Alba, A.E.; Rubio, M.B.; Hermosa, R.; Monte, E. Trichoderma and the plant heritable priming responses. J. Fungi 2021, 7, 318. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Mendoza, A.; Zaid, R.; Lawry, R.; Hermosa, R.; Monte, E.; Horwitz, B.A.; Mukherjee, P.K. Molecular dialogues between Trichoderma and roots: Role of the fungal secretome. Fungal Biol. Rev. 2018, 32, 62–85. [Google Scholar] [CrossRef]
- Ramírez-Valdespino, C.A.; Casas-Flores, S.; Olmedo-Monfil, V. Trichoderma as a model to study effector-like molecules. Front. Microbiol. 2019, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Ramírez, A.; Poveda, J.; Martín, J.I.; Hermosa, R.; Monte, E.; Nicolás, C. Salicylic acid prevents Trichoderma harzianum from entering the vascular system of the roots. Mol. Plant Pathol. 2014, 15, 823–831. [Google Scholar] [CrossRef]
- Hermosa, R.; Viterbo, A.; Chet, I.; Monte, E. Plant-beneficial effects of Trichoderma and of its genes. Microbiology 2012, 158, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Monte, E. The sophisticated evolution of Trichoderma to control insect pests. Proc. Natl. Acad. Sci. USA 2023, 120, e2301971120. [Google Scholar] [CrossRef]
- Carrero-Carrón, I.; Rubio, M.B.; Niño-Sánchez, J.; Navas, J.A.; Jiménez-Díaz, R.M.; Monte, E.; Hermosa, R. Interactions between Trichoderma harzianum and defoliating Verticillium dahliae in resistant and susceptible wild olive clones. Plant Pathol. 2018, 67, 1758–1767. [Google Scholar] [CrossRef]
- Giraldo, M.; Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Microbiol. 2013, 11, 800–814. [Google Scholar] [CrossRef]
- Lo Presti, L.; Kahmann, R. How filamentous plant pathogen effectors are translocated to host cells. Curr. Opin. Plant Biol. 2017, 38, 19–24. [Google Scholar] [CrossRef]
- Dodds, P.; Rathjen, J. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Ngou, B.P.M.; Ding, P.; Xin, X.F. PTI-ETI crosstalk: An integrative view of plant immunity. Curr. Opin. Plant Biol. 2021, 62, 102030. [Google Scholar] [CrossRef] [PubMed]
- Ngou, B.P.M.; Jones, J.D.G.; Ding, P. Plant immune networks. Trends Plant Sci. 2022, 27, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Zavaleta, C.Y.; García-Barrera, L.J.; Rodríguez-Verástegui, L.L.; Arrieta-Flores, D.; Gregorio-Jorge, J. An overview of PRR- and NLR-mediated immunities: Conserved signaling components across the plant kingdom that communicate both pathways. Int. J. Mol. Sci. 2022, 23, 12974. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gomollon, S.; Baulcombe, D.C. Roles of RNA silencing in viral and non-viral plant immunity and in the crosstalk between disease resistance systems. Nat. Rev. Mol. Cell Biol. 2022, 23, 645–662. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Wang, H.; Hu, P.; Hamby, R.; Jin, H. Small RNAs-big players in plant-microbe interactions. Cell Cross Microbe 2019, 26, 173–182. [Google Scholar] [CrossRef]
- Katiyar-Agarwal, S.; Jin, H. Role of small RNAs in host-microbe interactions. Annu. Rev. Phytopathol. 2010, 48, 225–246. [Google Scholar] [CrossRef]
- He, X.F.; Fang, Y.Y.; Feng, L.; Guo, H.S. Characterization of conserved and novel microRNAs and their targets, including a TuMV-induced TIR-NBS-LRR class R gene-derived novel miRNA in Brassica. FEBS Lett. 2008, 582, 2445–2452. [Google Scholar] [CrossRef]
- Fei, Q.; Yang, L.; Liang, W.; Zhang, D.; Meyers, B.C. Dynamic changes of small RNAs in rice spikelet development reveal specialized reproductive phasiRNA pathways. J. Exp. Bot. 2016, 67, 6037–6049. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Vaucheret, H. Plant argonautes. Trends Plant Sci. 2008, 13, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. microRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. microRNAs and their regulatory roles in plant–environment interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef]
- Vazquez, F.; Blevins, T.; Ailhas, J.; Boller, T.; Meins, F., Jr. Evolution of Arabidopsis MIR genes generates novel microRNA classes. Nucleic Acids Res. 2008, 36, 6429–6438. [Google Scholar] [CrossRef]
- Chávez Montes, R.A.; Rosas-Cárdenas, F.D.F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, N.; Meyers, B.C.; Green, P.J.; de Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef]
- Liu, Y.; Teng, C.; Xia, R.; Meyers, B.C. PhasiRNAs in plants: Their biogenesis, genic sources, and roles in stress responses, development, and reproduction. Plant Cell 2020, 32, 3059–3080. [Google Scholar] [CrossRef]
- Liu, N.; Avramova, Z. Molecular mechanism of the priming by jasmonic acid of specific dehydration stress response genes in Arabidopsis. Epigenetics Chromatin. 2016, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Marra, R.; Ambrosino, P.; Carbone, V.; Vinale, F.; Woo, S.L.; Ruocco, M.; Ciliento, R.; Lanzuise, S.; Ferraioli, S.; Soriente, I.; et al. Study of the three-way interaction between Trichoderma atroviride, plant and fungal pathogens by using a proteomic approach. Curr. Genet. 2006, 50, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Shoresh, M.; Harman, G.E. The molecular basis of shoot responses of maize seedlings to Trichoderma harzianum T22 inoculation of the root: A proteomic approach. Plant Physiol. 2008, 147, 2147–2163. [Google Scholar] [CrossRef] [PubMed]
- Manganiello, G.; Sacco, A.; Ercolano, M.R.; Vinale, F.; Lanzuise, S.; Pascale, A.; Napolitano, M.; Lombardi, N.; Lorito, M.; Woo, S.L. Modulation of tomato response to Rhizoctonia solani by Trichoderma harzianum and its secondary metabolite harzianic acid. Front. Microbiol. 2018, 9, 1966. [Google Scholar] [CrossRef] [PubMed]
- Salamon, S.; Żok, J.; Gromadzka, K.; Błaszczyk, L. Expression patterns of miR398, miR167, and miR159 in the interaction between bread wheat (Triticum aestivum L.) and pathogenic Fusarium culmorum and beneficial Trichoderma fungi. Pathogens 2021, 10, 1461. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo-Prudencio, O.G.; Estrada-Rivera, M.; Dautt-Castro, M.; Arteaga-Vazquez, M.A.; Arenas-Huertero, C.; Rosendo-Vargas, M.M.; Jin, H.; Casas-Flores, S. The small RNA-mediated gene silencing machinery is required in Arabidopsis for stimulation of growth, systemic disease resistance, and suppression of the nitrile-specifier gene NSP4 by Trichoderma atroviride. Plant J. 2022, 109, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, X.; Chen, J. Identification of miRNAs involved in maize-induced systemic resistance primed by Trichoderma harzianum T28 against Cochliobolus heterostrophus. J. Fungi 2023, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Medina, A.; Fernandez, I.; Lok, G.B.; Pozo, M.J.; Pieterse, C.M.; Van Wees, S.C. Shifting from priming of salicylic acid- to jasmonic acid-regulated defences by Trichoderma protects tomato against the root knot nematode Meloidogyne incognita. New Phytol. 2017, 213, 1363–1377. [Google Scholar] [CrossRef]
- Medeiros, H.A.; Araújo Filho, J.V.; Freitas, L.G.; Castillo, P.; Rubio, M.B.; Hermosa, R.; Monte, E. Tomato progeny inherit resistance to the nematode Meloidogyne javanica linked to plant growth induced by the biocontrol fungus Trichoderma atroviride. Sci. Rep. 2017, 7, 40216. [Google Scholar] [CrossRef]
- Domínguez, S.; Rubio, M.B.; Cardoza, R.E.; Gutiérrez, S.; Nicolás, C.; Bettiol, W.; Hermosa, R.; Monte, E. Nitrogen metabolism and growth enhancement in tomato plants challenged with Trichoderma harzianum expressing the Aspergillus nidulans acetamidase amdS gene. Front. Microbiol. 2016, 7, 1182. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.B.; Hermosa, R.; Vicente, R.; Gómez-Acosta, F.A.; Morcuende, R.; Monte, E.; Bettiol, W. The combination of Trichoderma harzianum and chemical fertilization leads to the deregulation of phytohormone networking, preventing the adaptative responses of tomato plants to salt stress. Front. Plant Sci. 2017, 8, 294. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shen, H.; Yuan, S.; Dai, X.; Yang, C. miRNAs and lncRNAs in tomato: Roles in biotic and abiotic stress responses. Front. Plant Sci. 2023, 13, 1094459. [Google Scholar] [CrossRef] [PubMed]
- Kamble, M.V.; Shahapurkar, A.B.; Adhikari, S.; Geetha, N.; Syed, A.; Ahmed, B.; Jogaiah, S. Identification and characterization of downy mildew-responsive microRNAs in Indian Vitis vinifera by high-throughput sequencing. J. Fungi 2021, 7, 899. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.R.; Kazan, K.; Manners, J.M. Exploiting pathogens’ tricks of the trade for engineering of plant disease resistance: Challenges and opportunities. Microb. Biotechnol. 2013, 6, 212–222. [Google Scholar] [CrossRef]
- Holt, D.B.; Gupta, V.; Meyer, D.; Abel, N.B.; Andersen, S.U.; Stougaard, J.; Markmann, K. micro RNA 172 (miR172) signals epidermal infection and is expressed in cells primed for bacterial invasion in Lotus japonicus roots and nodules. New Phytologist. 2015, 208, 241–256. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef]
- Jeong, D.H.; Park, S.; Zhai, J.; Gurazada, S.G.R.; De Paoli, E.; Meyers, B.C.; Green, P.J. Massive analysis of rice small RNAs: Mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23, 4185–4207. [Google Scholar] [CrossRef]
- Niu, C.; Li, H.; Jiang, L.; Yan, M.; Li, C.; Geng, D.; Xie, Y.; Yan, Y.; Shen, X.; Chen, P.; et al. Genome-wide identification of drought-responsive microRNAs in two sets of Malus from interspecific hybrid progenies. Hortic. Res. 2019, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Cui, J.; Zhai, J.; Li, J.; Han, L.; Meng, J. High-throughput sequencing reveals differential expression of miRNAs in tomato inoculated with Phytophthora infestans. Planta 2015, 241, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- López-Galiano, M.J.; Sentandreu, V.; Martínez-Ramírez, A.C.; Rausell, C.; Real, M.D.; Camañes, G.; Ruiz-Rivero, O.; Crespo-Salvador, O.; García-Robles, I. Identification of stress associated microRNAs in Solanum lycopersicum by high-throughput sequencing. Genes 2019, 10, 475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ji, H.M.; Gao, Y.; Cao, X.X.; Mao, H.Y.; Ouyang, S.Q.; Liu, P. An integrated analysis of mRNA and sRNA transcriptional profiles in tomato root: Insights on tomato wilt disease. PLoS ONE 2018, 13, e0206765. [Google Scholar] [CrossRef] [PubMed]
- Baldrich, P.; Bélanger, S.; Kong, S.; Pokhrel, S.; Tamim, S.; Teng, C.; Schiebout, C.; Gurazada, S.G.R.; Gupta, P.; Patel, P.; et al. The evolutionary history of small RNAs in Solanaceae. Plant Physiol. 2022, 189, 644–665. [Google Scholar] [CrossRef] [PubMed]
- Arazi, T.; Khedia, J. Tomato microRNAs and their functions. Int. J. Mol. Sci. 2022, 23, 11979. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, S.; Verma, R.; Lata, C.; Sanyal, I.; Rai, S.P. microRNA 166: An evolutionarily conserved stress biomarker in land plants targeting HD-ZIP family. Physiol. Mol. Biol. Plants 2021, 27, 2471–2485. [Google Scholar] [CrossRef]
- Cui, J.; Jiang, N.; Hou, X.; Wu, S.; Zhang, Q.; Meng, J.; Luan, Y. Genome-wide identification of lncRNAs and analysis of ceRNA networks during tomato resistance to Phytophthora infestans. Phytopathology 2020, 110, 456–464. [Google Scholar] [CrossRef]
- Clepet, C.; Devani, R.S.; Boumlik, R.; Hao, Y.; Morin, H.; Marcel, F.; Verdenaud, M.; Mania, B.; Brisou, G.; Citerne, S.; et al. The miR166-SlHB15A regulatory module controls ovule development and parthenocarpic fruit set under adverse temperatures in tomato. Mol. Plant 2021, 14, 1185–1198. [Google Scholar] [CrossRef]
- Prasad, A.; Prasad, M. Host-virus interactions mediated by long non-coding RNAs. Virus Res. 2021, 298, 198402. [Google Scholar] [CrossRef]
- Wang, J.; Yu, W.; Yang, Y.; Li, X.; Chen, T.; Liu, T.; Ma, N.; Yang, X.; Liu, R.; Zhang, B. Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci. Rep. 2015, 5, 16946. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, J.; Cao, M.; Gao, X.; Wang, D.; Liu, B.; Chen, Q. Genome-wide identification and characterization of HD-ZIP genes in potato. Gene 2019, 697, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.L.; Suzuki, R.; Cabrera, J.; Nakagami, S.; Sagara, T.; Ejima, C.; Sano, R.; Aoki, Y.; Olmo, R.; Kurata, T. Root-knot and cyst nematodes activate procambium-associated genes in Arabidopsis roots. Front. Plant Sci. 2017, 8, 1195. [Google Scholar] [CrossRef] [PubMed]
- Elhiti, M.; Stasolla, C. Structure and function of homodomain-leucine zipper (HD-Zip) proteins. Plant Signal. Behav. 2009, 4, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, Q.; Zuo, Z.-F.; Liu, L. microRNA398: A master regulator of plant development and stress responses. Int. J. Mol. Sci. 2022, 23, 10803. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Wang, W.; Liu, P. Identification and functional analysis of novel and conserved microRNAs in tomato. Mol. Biol. Rep. 2014, 41, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Candar-Cakir, B.; Arican, E.; Zhang, B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Gao, C.; Zhang, H.; Wen, F.; Tao, L.; Fu, G.; Xiong, J. The evolution and functional roles of miR408 and its targets in plants. Int. J. Mol. Sci. 2022, 23, 530. [Google Scholar] [CrossRef]
- Naqvi, A.R.; Haq, Q.M.; Mukherjee, S.K. microRNA profiling of tomato leaf curl new Delhi virus (tolcndv) infected tomato leaves indicates that deregulation of mir159/319 and mir172 might be linked with leaf curl disease. Virol. J. 2010, 7, 281. [Google Scholar] [CrossRef]
- Li, F.; Pignatta, D.; Bendix, C.; Brunkard, J.O.; Cohn, M.M.; Tung, J.; Sun, H.; Kumar, P.; Baker, B. microRNA regulation of plant innate immune receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1790–1795. [Google Scholar] [CrossRef]
- Seo, E.; Kim, T.; Park, J.H.; Yeom, S.I.; Kim, S.; Seo, M.K.; Shin, C.; Choi, D. Genome-wide comparative analysis in Solanaceous species reveals evolution of microRNAs targeting defense genes in Capsicum spp. DNA Res. 2018, 25, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.A.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Park, G.; Atamian, H.S.; Han, C.S.; Stajich, J.E.; Kaloshian, I.; Borkovich, K.A. microRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathog. 2014, 10, e1004464. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wu, F. Characterization of miRNAs associated with Botrytis cinerea infection of tomato leaves. BMC Plant Biol. 2015, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sarkar, A.; Chowdhury, S.; Singh, R.; Mukherjee, A.; Ghosh, Z.; Kundu, P. Heightened miR6024-NLR interactions facilitate necrotrophic pathogenesis in tomato. Plant Mol. Biol. 2022, 109, 717–739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Paasch, B.C.; Chen, J.; Day, B.; He, S.Y. An important role of l-fucose biosynthesis and protein fucosylation genes in Arabidopsis immunity. New Phytol. 2019, 222, 981–994. [Google Scholar] [CrossRef] [PubMed]
- FastQC: A Quality Control Tool for High throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 May 2021).
- Bushnell, B.; Rood, J.; Singer, E. BBMerge–accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, 0185056. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, Y. miR-PREFeR: An accurate, fast and easy-to-use plant miRNA prediction tool using small RNA-Seq data. Bioinformatics 2014, 30, 2837–2839. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing v3.6.1. R Foundation for Statistical Computing; R Core Team: Vienna, Austria, 2019. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate- A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.7. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 14 July 2023).
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. Available online: https://cran.r-project.org/web/packages/ggpubr/index.html (accessed on 28 September 2023).
- Gao, C.H.; Yu, G.; Cai, P. ggVennDiagram: An intuitive, easy-to-use, and highly customizable R package to generate Venn diagram. Front. Genet. 2021, 12, 706907. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version Version 1, 726. 2012. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 30 September 2023).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Library | Biological Replicate | Number of Raw Reads | Number of Reads after QC | Overall Alignment Rate (%) against S. lycopersicum Genome (from QC Reads) | Number of Reads That Aligned Once against S. lycopersicum Genome | Number of Reads Aligned against miRNA Candidates |
|---|---|---|---|---|---|---|
| 1 | C_R1 | 31,588,579 | 30,037,348 | 98.5 | 6,491,685 | 692,721 |
| 2 | C_R2 | 18,479,108 | 17,586,087 | 98.4 | 4,280,339 | 315,717 |
| 3 | C_R3 | 32,775,529 | 31,349,341 | 95.6 | 6,584,081 | 539,053 |
| 4 | T11_R1 | 24,024,180 | 22,844,527 | 98.6 | 4,976,203 | 320,623 |
| 5 | T11_R2 | 17,523,164 | 16,525,822 | 98.1 | 5,066,436 | 278,130 |
| 6 | T11_R3 | 61,256,777 | 55,450,480 | 98.1 | 11,635,432 | 1,165,112 |
| miRNA_ID | miRNA_Name | MIR Family Name | Mean CPM Control | Mean CPM T11 | log2FC | lfcSE | padj (<0.1) |
|---|---|---|---|---|---|---|---|
| miRNA_1767 | novel miR1767 | MIR1767 | 0.46 | 0.00 | −6.243 | 1.673 | 0.00296 |
| miRNA_237 | miR408 | MIR408 | 2.36 | 0.67 | −1.896 | 0.478 | 0.00141 |
| miRNA_2072 | miR398-3p | MIR398 | 2.55 | 0.78 | −1.745 | 0.679 | 0.06613 |
| miRNA_965 | miR166a | MIR166 | 5901.95 | 3597.31 | −0.805 | 0.145 | 1.99 × 10−6 |
| miRNA_1734 | miR6027-5p | MIR6027 | 185.79 | 141.93 | −0.471 | 0.091 | 6.66 × 10−6 |
| miRNA_1908 | miR9471b-3p | MIR9741 | 57.08 | 83.67 | 0.428 | 0.142 | 0.02008 |
| miRNA_607 | miR5300 | MIR5300 | 17.26 | 25.20 | 0.454 | 0.132 | 0.00553 |
| miRNA_608 | miR5300 | 17.26 | 25.20 | 0.454 | 0.132 | 0.00553 | |
| miRNA_181 | miR6024 | MIR6024 | 4.08 | 7.96 | 0.856 | 0.245 | 0.00553 |
| miRNA_257 | novel miR257 | MIR257 | 0.49 | 1.25 | 1.169 | 0.432 | 0.04789 |
| miRNA_275 | novel miR275 | MIR275 | 0.02 | 0.00 | 10.315 | 3.307 | 0.01574 |
| miRNA ID | miRNA Name | miRNA Mature Sequence | Length | % GC | S. lycopersicum Chromosome | miRNA Chromosome Start Position | miRNA Chromosome End Position | Strand | Minimum Free Energy (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|
| miRNA_1767 | novel miR1767 | CUUCAACUUUGGGUGUGCACAAGU | 24 | 45.83% | 11 | 2,825,245 | 2,825,268 | - | −61.8 |
| miRNA_257 | novel miR257 | AAAGAGAUUUUGAACUUGAGACCU | 24 | 33.33% | 1 | 88,918,167 | 88,918,190 | - | −24.1 |
| miRNA_275 | novel miR275 | CUCUGAGAUUUCGGGCAUAGGUU | 23 | 47.83% | 2 | 19,634,044 | 19,634,334 | - | −222.5 |
| miRNA | Target Accession | Exp | UPE | miRNA Length | Target Start | Target End | miRNA Aligned Fragment | Target Aligned Fragment | Inhibition | KEGG Orthologue ID | Target Description |
|---|---|---|---|---|---|---|---|---|---|---|---|
| miR166a | Solyc11g069470.3.1 | 1 | 23.445 | 21 | 653 | 673 | UCGGACCAGGCUUCAUUCCCC | CUGGGAUGAAGCCUGGUCCGG | Cleavage | K09338 | Class III homeodomain-leucine zipper |
| Solyc10g006720.4.1 | 3.5 | 24.431 | 21 | 551 | 571 | UCGGACCAGGCUUCAUUCCCC | CUGGAAUGAAGCUUGGGCGGA | Cleavage | K04733 | G-type lectin S-receptor-like serine/threonine-protein kinase | |
| Solyc03g121640.3.1 | 3.5 | 13.824 | 21 | 795 | 814 | UCGGACCAGGCUUCAUUCCCC | AAGGAAUGAAGCUUGG-CCGA | Cleavage | K04077 | Chaperonin-60 kDa protein | |
| miR6027-5p | Solyc07g047990.1.1 | 2 | 16.738 | 22 | 435 | 456 | AUGGGUAGCACAAGGAUUAAUG | UCAUGAUCCUUGUGUUAUUCAU | Cleavage | K08867 | MAP kinase kinase kinase 49 |
| Solyc09g064270.3.1 | 3 | 12.926 | 22 | 1783 | 1804 | AUGGGUAGCACAAGGAUUAAUG | UUCUAAUCCUCGUGUUAUUCAU | Cleavage | K13430 | Receptor-like serine/threonine-protein kinase ALE2 | |
| Solyc01g107670.2.1 | 3.5 | 14.541 | 22 | 213 | 234 | AUGGGUAGCACAAGGAUUAAUG | CUCUGUUCCUCGUGUUACCCAU | Cleavage | NA | Leucine-rich repeat receptor-like protein kinase | |
| miR398-3p | Solyc02g078720.4.1 | 3.5 | 16.291 | 21 | 652 | 672 | UGUGUUCUCAGGUUACCCCUG | AAAGGGUAACCUGAGCAUAUA | Cleavage | NA | Multidrug resistance protein |
| Solyc05g006630.4.1 | 3.5 | 22.484 | 21 | 558 | 578 | UGUGUUCUCAGGUUACCCCUG | CUGGGGAAACUUGAUAAUACA | Cleavage | K19613 | Disease-resistance-like protein (TIR-NBS-LRR class) | |
| novel miR1767 | Solyc12g014490.3.1 | 2.5 | 15.25 | 24 | 1393 | 1416 | CUUCAACUUUGGGUGUGCACAAGU | AGAGGUGCACACUUAAAUUUGAAG | Cleavage | K16732 | Microtubule-associated protein MAP65-1c |
| Solyc03g116760.3.1 | 3 | 19.108 | 24 | 1251 | 1274 | CUUCAACUUUGGGUGUGCACAAGU | GAACUUGCAGACCCAAGGUUGAGU | Cleavage | K13416 | LRR receptor-like serine/threonine-protein kinase FEI 1 | |
| Solyc05g053260.3.1 | 3.5 | 18.48 | 24 | 199 | 222 | CUUCAACUUUGGGUGUGCACAAGU | GUGGAAUCAUGCCUAAAGUUGAAG | Cleavage | NA | DNA (Cytosine-5)-methyltransferase DRM2 | |
| miR5300 | Solyc03g116550.4.1 | 2.5 | 17.866 | 22 | 936 | 957 | UCCCCAGUCCAGGCAUUCCAAC | ACAGGAAACCUUGGACUGGGGA | Cleavage | NA | O-fucosyltransferase family protein (AT1G52630-like protein) |
| Solyc05g008650.1.1 | 3 | 20.456 | 22 | 1282 | 1303 | UCCCCAGUCCAGGCAUUCCAAC | GUUGGAAUGCCUGGACUUGGCA | Cleavage | K13453 | Late blight-resistance protein R1-A (NBS-coding resistance gene protein) | |
| Solyc06g064690.2.1 | 3 | 19.297 | 22 | 40 | 61 | UCCCCAGUCCAGGCAUUCCAAC | UAUGGAAUGCCUGGACUUGGUA | Cleavage | K13453 | NBS-coding resistance gene analog | |
| miR6024 | Solyc10g051050.3.1 | 1 | 21.519 | 22 | 665 | 686 | UUUUAGCAAGAGUUGUUUUACC | GGUAAGACAACUCUUGCUAGAA | Cleavage | K13453 | Disease-resistance protein (AT4G27190-like protein) |
| Solyc11g065780.3.1 | 2.5 | 14.182 | 22 | 469 | 490 | UUUUAGCAAGAGUUGUUUUACC | GGUAAGACAACACUUGCUAAAG | Translation | K15078 | CC-NBS-LRR type resistance-like protein/Cc-nbs-resistance protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olmo, R.; Quijada, N.M.; Morán-Diez, M.E.; Hermosa, R.; Monte, E. Identification of Tomato microRNAs in Late Response to Trichoderma atroviride. Int. J. Mol. Sci. 2024, 25, 1617. https://doi.org/10.3390/ijms25031617
Olmo R, Quijada NM, Morán-Diez ME, Hermosa R, Monte E. Identification of Tomato microRNAs in Late Response to Trichoderma atroviride. International Journal of Molecular Sciences. 2024; 25(3):1617. https://doi.org/10.3390/ijms25031617
Chicago/Turabian StyleOlmo, Rocío, Narciso M. Quijada, María Eugenia Morán-Diez, Rosa Hermosa, and Enrique Monte. 2024. "Identification of Tomato microRNAs in Late Response to Trichoderma atroviride" International Journal of Molecular Sciences 25, no. 3: 1617. https://doi.org/10.3390/ijms25031617