
The Role of Bond Functions in Describing Intermolecular Electron Correlation for Van der Waals Dimers: A Study of (CH4)2 and Ne2

Abstract
:1. Introduction
2. Results and Discussion
2.1. Theory
2.1.1. Reduced Density Matrices in the CCSD Method
2.1.2. Cusp Region
2.2. Computational Details
2.2.1. Constructing the Two-Particle Density Matrix
2.2.2. Details of the Calculations
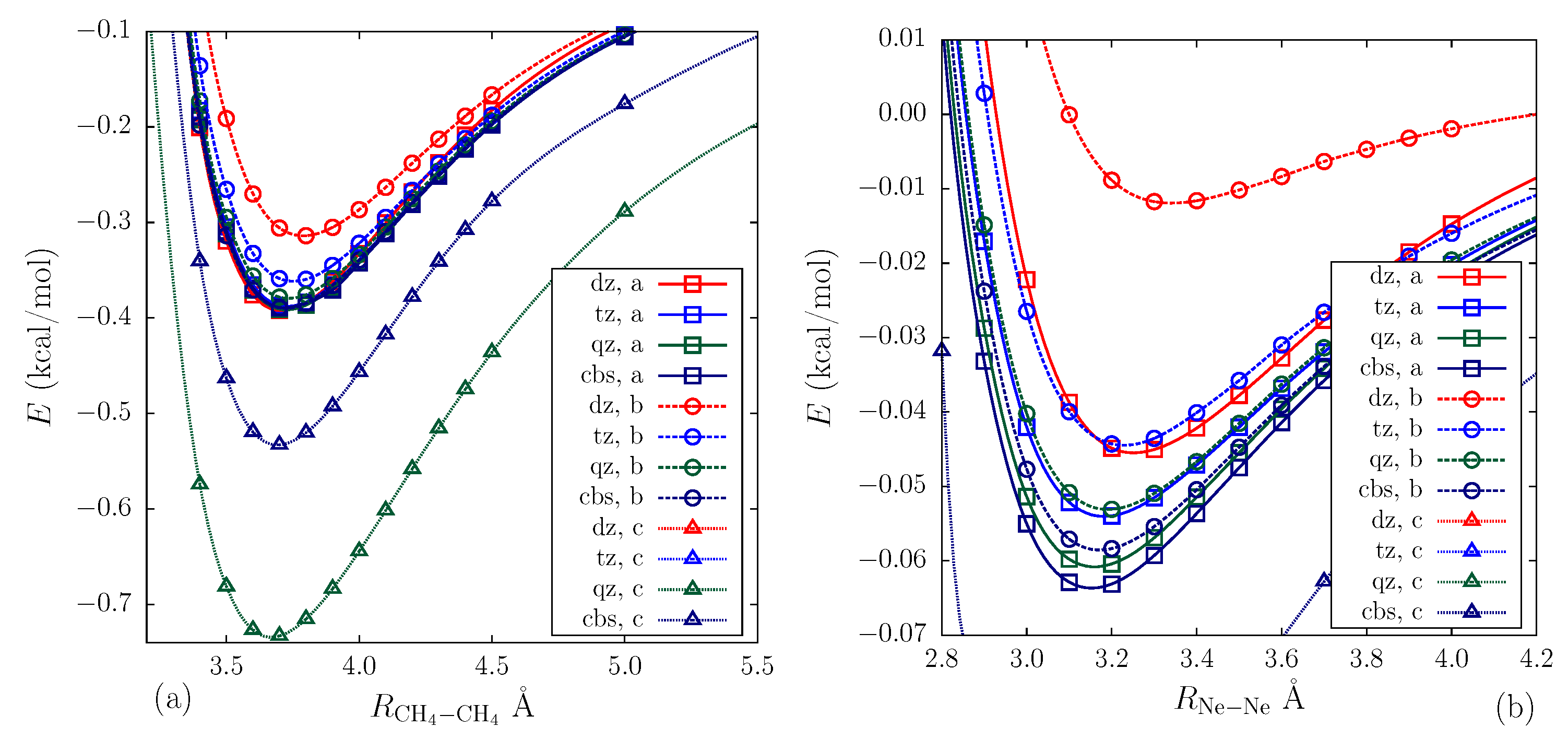
2.3. Calculation Results
3. Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BSSE | Basis set superposition error |
| 2-RDM | Two-particle reduced density matrix |
| PES | Potential energy surface |
| CCSD | Coupled cluster with single and double excitations |
| GMRES | Generalized minimal residual method |
| DIIS | Direct inversion in the iterative subspace |
| CCSD-RDM | Coupled-cluster singles and doubles with reduced density matrices |
References
- Hobza, P.; Šponer, J. Toward True DNA Base-Stacking Energies: MP2, CCSD(T), and Complete Basis Set Calculations. J. Am. Chem. Soc. 2002, 124, 11802–11808. [Google Scholar] [CrossRef]
- Danilov, V.I.; Anisimov, V.M.; Kurita, N.; Hovorun, D. MP2 and DFT studies of the DNA rare base pairs: The molecular mechanism of the spontaneous substitution mutations conditioned by tautomerism of bases. Chem. Phys. Lett. 2005, 412, 285–293. [Google Scholar] [CrossRef]
- Chung, G.; Oh, H.; Lee, D. Tautomerism and isomerism of guanine–cytosine DNA base pair: Ab initio and density functional theory approaches. J. Mol. Struct. THEOCHEM 2005, 730, 241–249. [Google Scholar] [CrossRef]
- Yildirim, I.; Turner, D.H. RNA Challenges for Computational Chemists. Biochemistry 2005, 44, 13225–13234. [Google Scholar] [CrossRef]
- Cysewski, P.; Czyżnikowska-Balcerak, Ż. The MP2 quantum chemistry study on the local minima of guanine stacked with all four nucleic acid bases in conformations corresponding to mean B-DNA. J. Mol. Struct. THEOCHEM 2005, 757, 29–36. [Google Scholar] [CrossRef]
- Jurečka, P.; Šponer, J.; Černý, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef]
- Priyakumar, U.D.; MacKerell, A.D. Base Flipping in a GCGC Containing DNA Dodecamer: A Comparative Study of the Performance of the Nucleic Acid Force Fields, CHARMM, AMBER, and BMS. J. Chem. Theory Comput. 2005, 2, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Langner, K.M.; Kedzierski, P.; Sokalski, W.A.; Leszczynski, J. Physical Nature of Ethidium and Proflavine Interactions with Nucleic Acid Bases in the Intercalation Plane. J. Phys. Chem. B 2006, 110, 9720–9727. [Google Scholar] [CrossRef] [PubMed]
- Haley, T.P.; Graybill, E.R.; Cybulski, S.M. Ab Initio Calc. Dispers. Coefficients Nucleic Acid Base Pairs. J. Chem. Phys. 2006, 124, 204301. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, K.; Komeiji, Y.; Mochizuki, Y.; Kato, A.; Nakano, T.; Tanaka, S. Intra- and intermolecular interactions between cyclic-AMP receptor protein and DNA: Ab Initio Fragm. Mol. Orbital Study. J. Comput. Chem. 2006, 27, 948–960. [Google Scholar] [CrossRef]
- Cauët, E.; Liévin, J. Radical Cations of the Nucleic Bases and Radiation Damage to DNA: Ab Initio Study. In Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2007; pp. 121–147. [Google Scholar] [CrossRef]
- Šponer, J.; Riley, K.E.; Hobza, P. Nature and magnitude of aromatic stacking of nucleic acid bases. Phys. Chem. Chem. Phys. 2008, 10, 2595. [Google Scholar] [CrossRef]
- Rutledge, L.R.; Campbell-Verduyn, L.S.; Hunter, K.C.; Wetmore, S.D. Characterization of Nucleobase-Amino Acid Stacking Interactions Utilized by a DNA Repair Enzyme. J. Phys. Chem. B 2006, 110, 19652–19663. [Google Scholar] [CrossRef]
- Rutledge, L.R.; Durst, H.F.; Wetmore, S.D. Evidence for Stabilization of DNA/RNA-Protein Complexes Arising from Nucleobase-Amino Acid Stacking and T-Shaped Interactions. J. Chem. Theory Comput. 2009, 5, 1400–1410. [Google Scholar] [CrossRef]
- Butchosa, C.; Simon, S.; Voityuk, A.A. Conformational dependence of the electronic coupling for hole transfer between adenine and tryptophan. Comput. Theor. Chem. 2011, 975, 38–41. [Google Scholar] [CrossRef]
- Parrish, R.M.; Sherrill, C.D. Spatial assignment of symmetry adapted perturbation theory interaction energy components: The atomic SAPT partition. J. Chem. Phys. 2014, 141, 044115. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, F.; Dunivan, S.; Lao, K.U. Coupled cluster benchmarks of large noncovalent complexes: The L7 dataset as well as DNA–ellipticine and buckycatcher-fullerene. J. Chem. Phys. 2021, 154, 154104. [Google Scholar] [CrossRef]
- Kříž, K.; Řezáč, J. Benchmarking of Semiempirical Quantum-Mechanical Methods on Systems Relevant to Computer-Aided Drug Design. J. Chem. Inf. Model. 2020, 60, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Morawietz, T.; Artrith, N. Machine learning-accelerated quantum mechanics-based atomistic simulations for industrial applications. J. Comput.-Aided Mol. Des. 2020, 35, 557–586. [Google Scholar] [CrossRef]
- Villar, R.; Gil, M.J.; García, J.I.; Martínez-Merino, V. Are AM1 ligand-protein binding enthalpies good enough for use in the rational design of new drugs? J. Comput. Chem. 2005, 26, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Chałasiński, G.; Szczĕśniak, M.M. State of the Art and Challenges of the ab Initio Theory of Intermolecular Interactions. Chem. Rev. 2000, 100, 4227–4252. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.T.; Chao, S.D. Intermolecular potentials of the methane dimer calculated with Møller-Plesset perturbation theory and density functional theory. J. Chem. Phys. 2006, 125, 094312. [Google Scholar] [CrossRef] [PubMed]
- Rutskoy, B.V.; Bezrukov, D.S. Ab Initio Description of the Structure and Interaction Energy of Perhalomethane Dimers. Russ. J. Phys. Chem. A 2019, 93, 1519–1524. [Google Scholar] [CrossRef]
- Li, A.H.T.; Chao, S.D. Interaction energies of dispersion-bound methane dimer from coupled cluster method at complete basis set limit. J. Mol. Struct. THEOCHEM 2009, 897, 90–94. [Google Scholar] [CrossRef]
- Halkier, A.; Klopper, W.; Helgaker, T.; Jo/rgensen, P.; Taylor, P.R. Basis set convergence of the interaction energy of hydrogen-bonded complexes. J. Chem. Phys. 1999, 111, 9157–9167. [Google Scholar] [CrossRef]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-set convergence in correlated calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.C.M.; van Lenthe, J.H. State of the Art in Counterpoise Theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Dunning, T.H. A Road Map for the Calculation of Molecular Binding Energies. J. Phys. Chem. A 2000, 104, 9062–9080. [Google Scholar] [CrossRef]
- Burcl, R.; Chałasiński, G.; Bukowski, R.; Szczĕśniak, M.M. On the role of bond functions in interaction energy calculations: Ar···HCl, Ar···H2O, (HF)2. J. Chem. Phys. 1995, 103, 1498–1507. [Google Scholar] [CrossRef]
- Tecmer, P.; Boguslawski, K. Geminal-based electronic structure methods in quantum chemistry. Toward a geminal model chemistry. Phys. Chem. Chem. Phys. 2022, 24, 23026–23048. [Google Scholar] [CrossRef]
- Tao, F.M.; Pan, Y.K. Møller-Plesset perturbation investigation of the He2 potential and the role of midbond basis functions. J. Chem. Phys. 1992, 97, 4989–4995. [Google Scholar] [CrossRef]
- Tao, F.M.; Pan, Y.K. Ab Initio Potential Energy Curves Bind. Energies Ar2 Mg2. Mol. Phys. 1994, 81, 507–518. [Google Scholar] [CrossRef]
- Tao, F.M. The use of midbond functions for ab initio calculations of the asymmetric potentials of He–Ne and He–Ar. J. Chem. Phys. 1993, 98, 3049–3059. [Google Scholar] [CrossRef]
- Matveeva, R.; Falck Erichsen, M.; Koch, H.; Høyvik, I. The effect of midbond functions on interaction energies computed using MP2 and CCSD(T). J. Comput. Chem. 2021, 43, 121–131. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Describing Noncovalent Interactions beyond the Common Approximations: How Accurate Is the “Gold Standard,” CCSD(T) at the Complete Basis Set Limit? J. Chem. Theory Comput. 2013, 9, 2151–2155. [Google Scholar] [CrossRef]
- Řezáč, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef]
- Koch, H.; Fernández, B.; Christiansen, O. The benzene–argon complex: A ground and excited state ab initio study. J. Chem. Phys. 1998, 108, 2784–2790. [Google Scholar] [CrossRef]
- Partridge, H.; Bauschlicher, C.W. The dissociation energies of He2, HeH, and ArH: A bond function study. Mol. Phys. 1999, 96, 705–710. [Google Scholar] [CrossRef]
- Dutta, N.N.; Patkowski, K. Improving “Silver-Standard” Benchmark Interaction Energies with Bond Functions. J. Chem. Theory Comput. 2018, 14, 3053–3070. [Google Scholar] [CrossRef]
- Akin-Ojo, O.; Bukowski, R.; Szalewicz, K. Ab initio studies of He–HCCCN interaction. J. Chem. Phys. 2003, 119, 8379–8396. [Google Scholar] [CrossRef]
- Shaw, R.A.; Hill, J.G. Midbond basis functions for weakly bound complexes. Mol. Phys. 2018, 116, 1460–1470. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Rybak, S.; Szalewicz, K.; Jeziorski, B.; Jaszunski, M. Intraatomic correlation effects for the He–He dispersion and exchange/dispersion energies using explicitly correlated Gaussian geminals. J. Chem. Phys. 1987, 86, 5652–5659. [Google Scholar] [CrossRef]
- Korona, T. Second-order exchange-induction energy of intermolecular interactions from coupled cluster density matrices and their cumulants. Phys. Chem. Chem. Phys. 2008, 10, 6509. [Google Scholar] [CrossRef]
- Szalewicz, K.; Jeziorski, B. Symmetry-adapted double-perturbation analysis of intramolecular correlation effects in weak intermolecular interactions. Mol. Phys. 1979, 38, 191–208. [Google Scholar] [CrossRef]
- Baerends, E.J.; Gritsenko, O.V. A Quantum Chemical View of Density Functional Theory. J. Phys. Chem. A 1997, 101, 5383–5403. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F.; Bartlett, R.J. Coupled-cluster open-shell analytic gradients: Implementation of the direct product decomposition approach in energy gradient calculations. J. Chem. Phys. 1991, 95, 2623–2638. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F.; Bartlett, R.J. Analytic evaluation of energy gradients at the coupled-cluster singles and doubles level using quasi-restricted Hartree-Fock open-shell reference functions. J. Chem. Phys. 1991, 95, 2639–2645. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Watts, J.D.; Bartlett, R.J. A direct product decomposition approach for symmetry exploitation in many-body methods. I. Energy calculations. J. Chem. Phys. 1991, 94, 4334–4345. [Google Scholar] [CrossRef]
- Fitzgerald, G.; Harrison, R.J.; Bartlett, R.J. Analytic energy gradients for general coupled-cluster methods and fourth-order many-body perturbation theory. J. Chem. Phys. 1986, 85, 5143–5150. [Google Scholar] [CrossRef]
- Scheiner, A.C.; Scuseria, G.E.; Rice, J.E.; Lee, T.J.; Schaefer, H.F. Analytic evaluation of energy gradients for the single and double excitation coupled cluster (CCSD) wave function: Theory and application. J. Chem. Phys. 1987, 87, 5361–5373. [Google Scholar] [CrossRef]
- Lyakh, D.I.; Musiał, M.; Lotrich, V.F.; Bartlett, R.J. Multireference Nature of Chemistry: The Coupled-Cluster View. Chem. Rev. 2011, 112, 182–243. [Google Scholar] [CrossRef]
- Krylov, A.I. Equation-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker’s Guide to Fock Space. Annu. Rev. Phys. Chem. 2008, 59, 433–462. [Google Scholar] [CrossRef]
- Helgaker, T.; Jörgensen, P. Configuration-interaction energy derivatives in a fully variational formulation. Theor. Chim. Acta 1989, 75, 111–127. [Google Scholar] [CrossRef]
- Kato, T. On the eigenfunctions of many-particle systems in quantum mechanics. Commun. Pure Appl. Math. 1957, 10, 151–177. [Google Scholar] [CrossRef]
- Pan, X.Y.; Sahni, V. Integral coalescence conditions in D ⩾ 2 dimension space. J. Chem. Phys. 2003, 119, 7083–7086. [Google Scholar] [CrossRef]
- Kutzelnigg, W. r 12-Dependent terms in the wave function as closed sums of partial wave amplitudes for large l. Theor. Chim. Acta 1985, 68, 445–469. [Google Scholar] [CrossRef]
- Noga, J.; Kutzelnigg, W. Coupled cluster theory that takes care of the correlation cusp by inclusion of linear terms in the interelectronic coordinates. J. Chem. Phys. 1994, 101, 7738–7762. [Google Scholar] [CrossRef]
- Noga, J.; Kedžuch, S.; Šimunek, J.; Ten-no, S. Explicitly correlated coupled cluster F12 theory with single and double excitations. J. Chem. Phys. 2008, 128, 174103. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F. Analytic gradients for the coupled-cluster singles, doubles, and triples (CCSDT) model. J. Chem. Phys. 2002, 116, 1773–1782. [Google Scholar] [CrossRef]
- Taube, A.G.; Bartlett, R.J. Improving upon CCSD(T): ΛCCSD(T). I. Potential energy surfaces. J. Chem. Phys. 2008, 128, 044110. [Google Scholar] [CrossRef]
- Jo/rgensen, P.; Helgaker, T. Møller-Plesset energy derivatives. J. Chem. Phys. 1988, 89, 1560–1570. [Google Scholar] [CrossRef]
- Helgaker, T.; Jørgensen, P. Analytical Calculation of Geometrical Derivatives in Molecular Electronic Structure Theory. In Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 1988; pp. 183–245. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F. Coupled-cluster calculations of nuclear magnetic resonance chemical shifts. J. Chem. Phys. 1995, 103, 3561–3577. [Google Scholar] [CrossRef]
- Cybulski, S.M.; Toczyłowski, R.R. Ground state potential energy curves for He2, Ne2, Ar2, He–Ne, He–Ar, and Ne–Ar: A coupled-cluster study. J. Chem. Phys. 1999, 111, 10520–10528. [Google Scholar] [CrossRef]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Olsen, J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. [Google Scholar] [CrossRef]
- Tao, F.M. Bond functions, basis set superposition errors and other practical issues with ab initio calculations of intermolecular potentials. Int. Rev. Phys. Chem. 2001, 20, 617–643. [Google Scholar] [CrossRef]
- Coulson, C.A.; Neilson, A.H. Electron Correlation in the Ground State of Helium. Proc. Phys. Soc. 1961, 78, 831–837. [Google Scholar] [CrossRef]
- Boyd, R.J.; Yee, M.C. Angular aspects of electron correlation and the Coulomb hole. J. Chem. Phys. 1982, 77, 3578–3582. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2011, 2, 242–253. [Google Scholar] [CrossRef]
- Saad, Y.; Schultz, M.H. GMRES: A Generalized Minimal Residual Algorithm for Solving Nonsymmetric Linear Systems. SIAM J. Sci. Stat. Comput. 1986, 7, 856–869. [Google Scholar] [CrossRef]
- Yang, C.; Brabec, J.; Veis, L.; Williams-Young, D.B.; Kowalski, K. Solving Coupled Cluster Equations by the Newton Krylov Method. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Pulay, P. Convergence acceleration of iterative sequences. the case of SCF iteration. Chem. Phys. Lett. 1980, 73, 393–398. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Lee, T.J.; Schaefer, H.F. Accelerating the convergence of the coupled-cluster approach. Chem. Phys. Lett. 1986, 130, 236–239. [Google Scholar] [CrossRef]
- Ito, T.; Hayami, K. Preconditioned GMRES methods for least squares problems. Jpn. J. Ind. Appl. Math. 2008, 25, 185–207. [Google Scholar] [CrossRef]






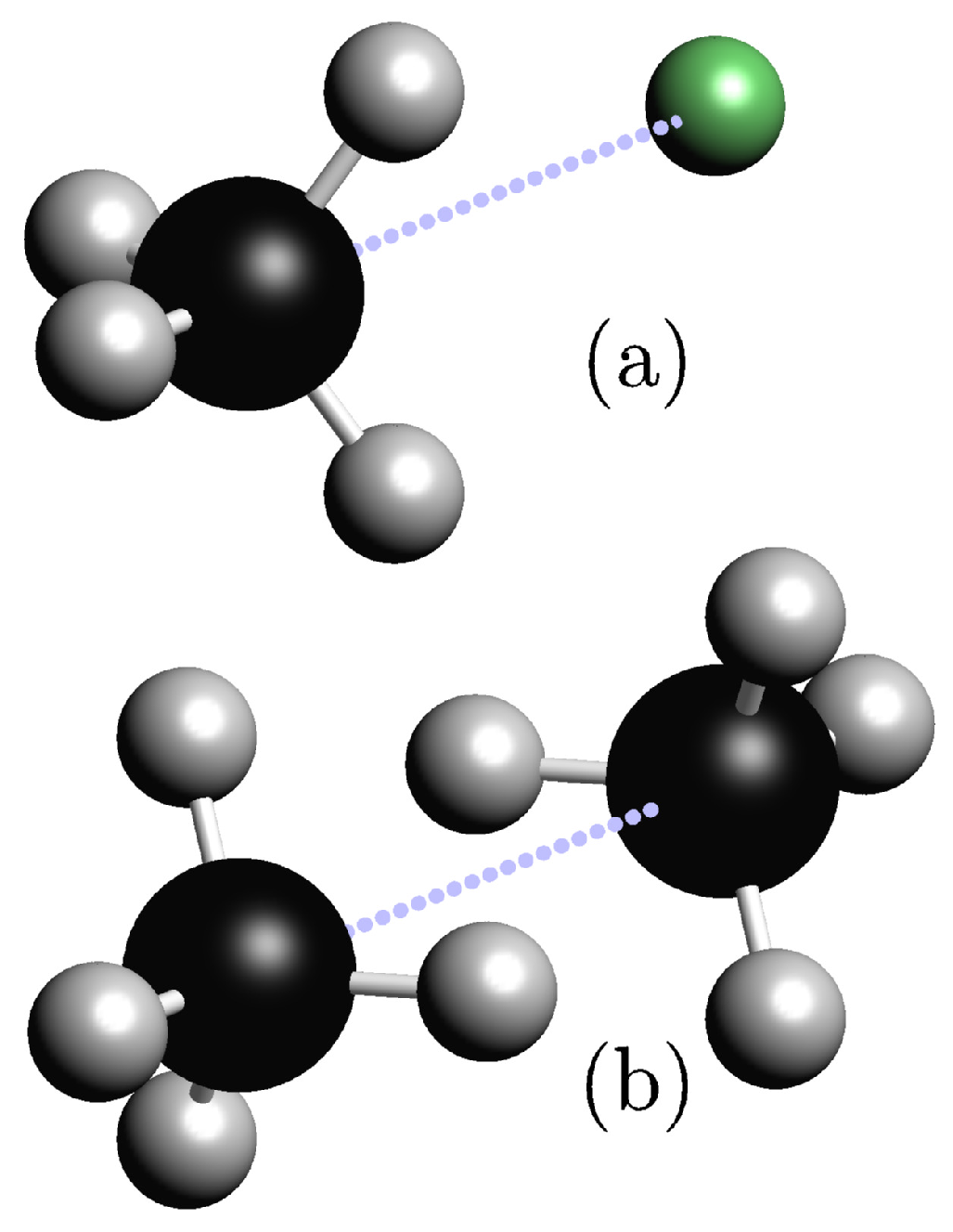{kind=link}
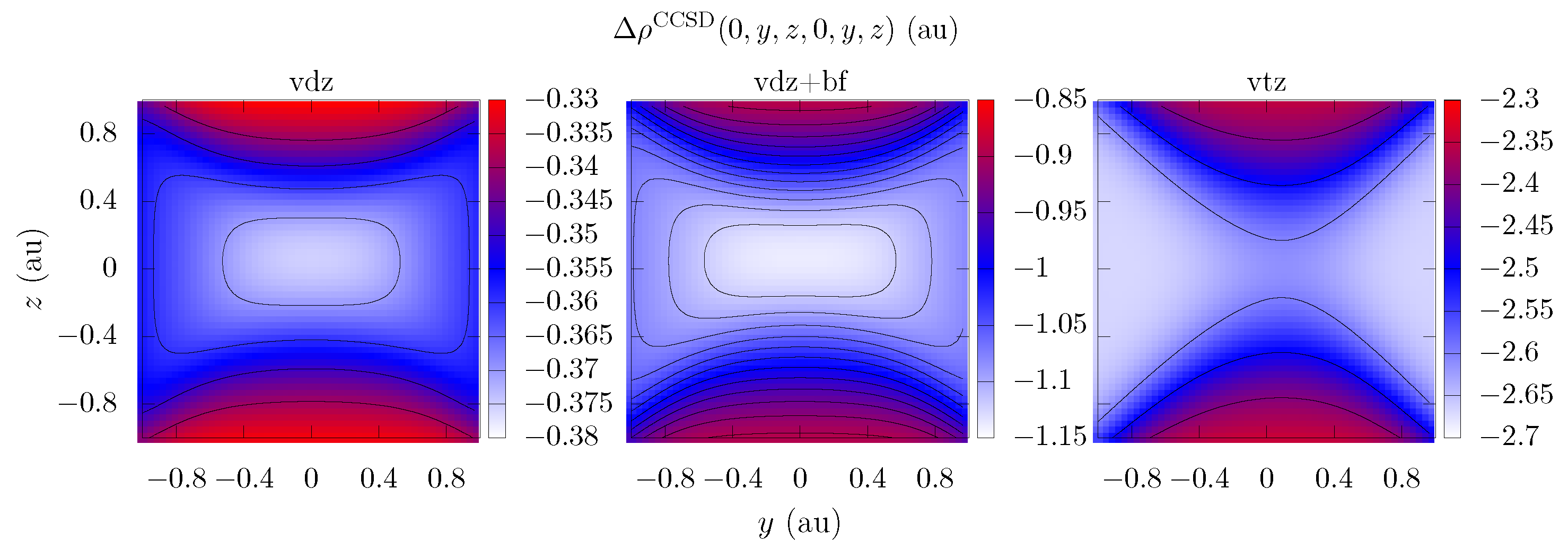{kind=link}
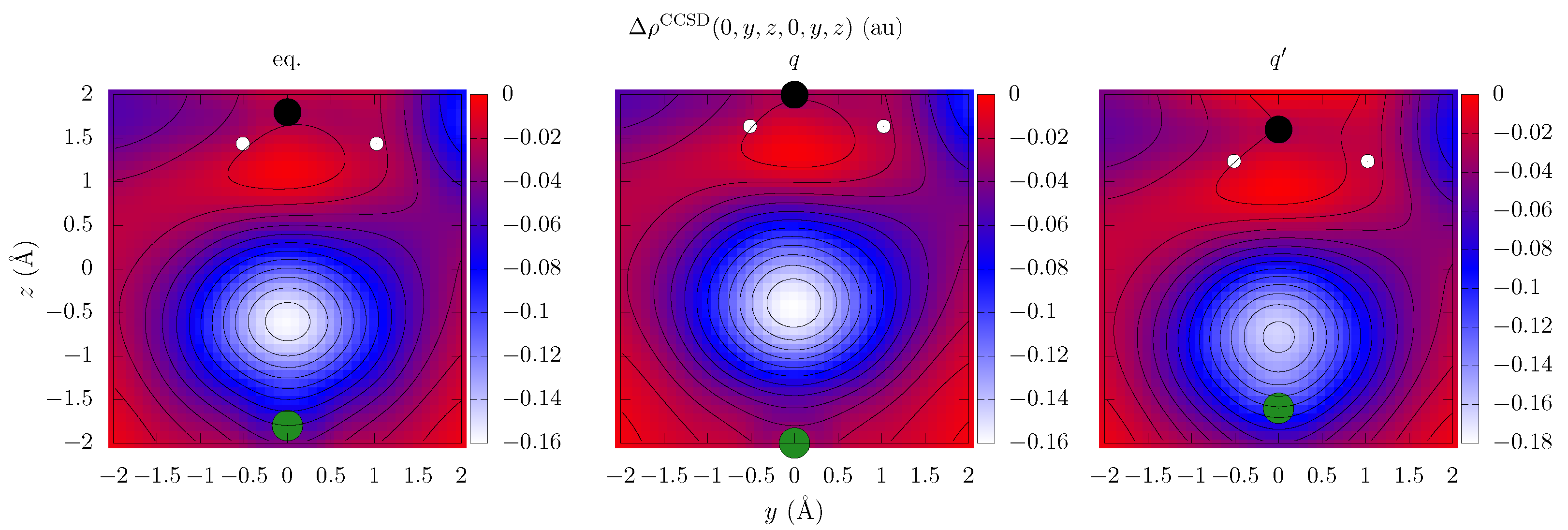{kind=link}
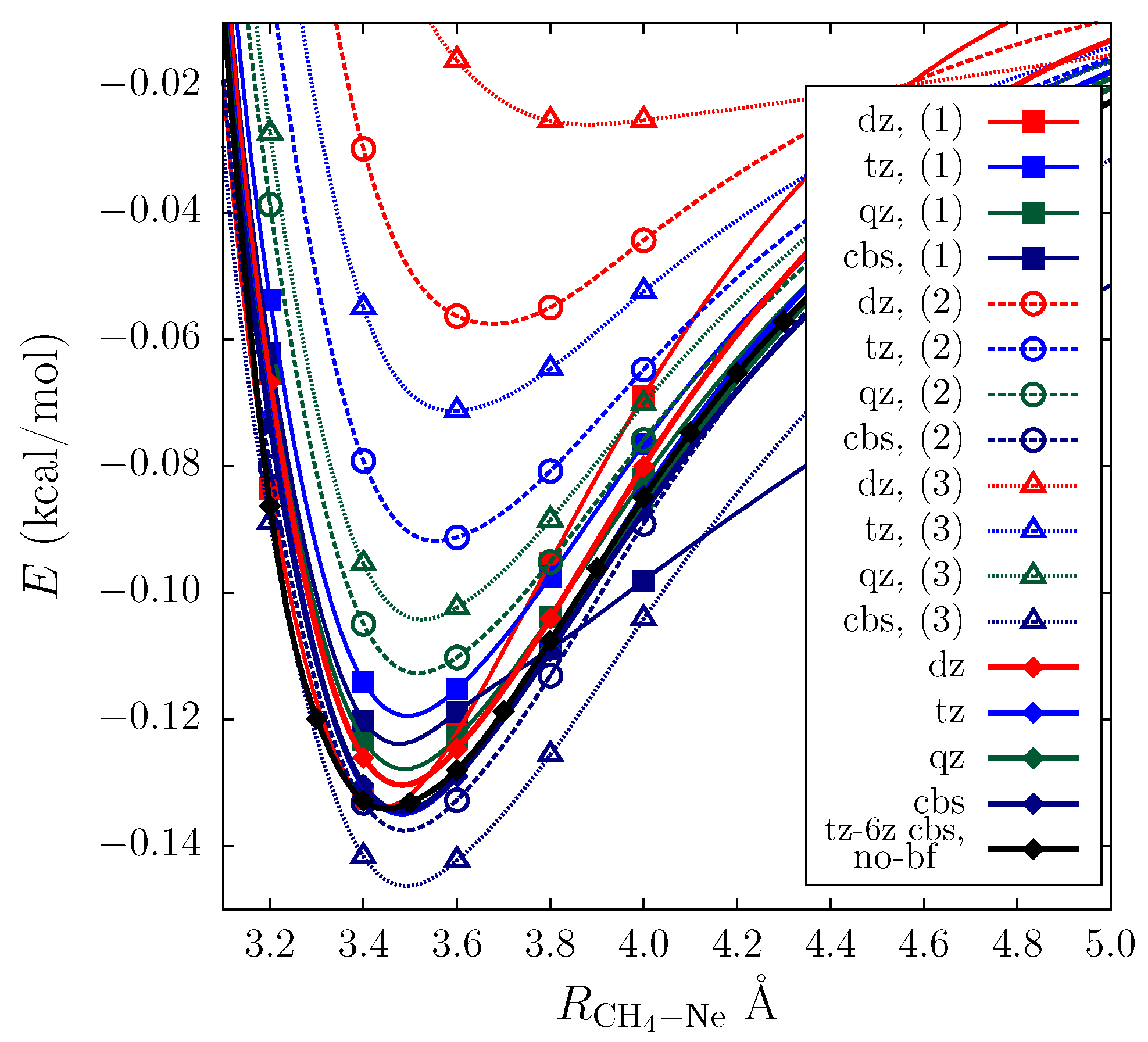{kind=link}
{kind=link}
| Electron Coordinates (a0) | cc-pVDZ | cc-pVTZ | cc-pVDZ +bf[33221] | ||
|---|---|---|---|---|---|
| x | y | z | |||
| 0 | 0 | 0 | −0.4143 | −2.71 | −1.26 |
| 0 | 0 | 0.9 | −0.321 | −2.66 | −0.808 |
| 0 | 0.9 | 0 | −0.355 | −2.33 | −1.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rutskoy, B.; Ozerov, G.; Bezrukov, D. The Role of Bond Functions in Describing Intermolecular Electron Correlation for Van der Waals Dimers: A Study of (CH4)2 and Ne2. Int. J. Mol. Sci. 2024, 25, 1472. https://doi.org/10.3390/ijms25031472
Rutskoy B, Ozerov G, Bezrukov D. The Role of Bond Functions in Describing Intermolecular Electron Correlation for Van der Waals Dimers: A Study of (CH4)2 and Ne2. International Journal of Molecular Sciences. 2024; 25(3):1472. https://doi.org/10.3390/ijms25031472
Chicago/Turabian StyleRutskoy, Bogdan, Georgiy Ozerov, and Dmitry Bezrukov. 2024. "The Role of Bond Functions in Describing Intermolecular Electron Correlation for Van der Waals Dimers: A Study of (CH4)2 and Ne2" International Journal of Molecular Sciences 25, no. 3: 1472. https://doi.org/10.3390/ijms25031472