
Splicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
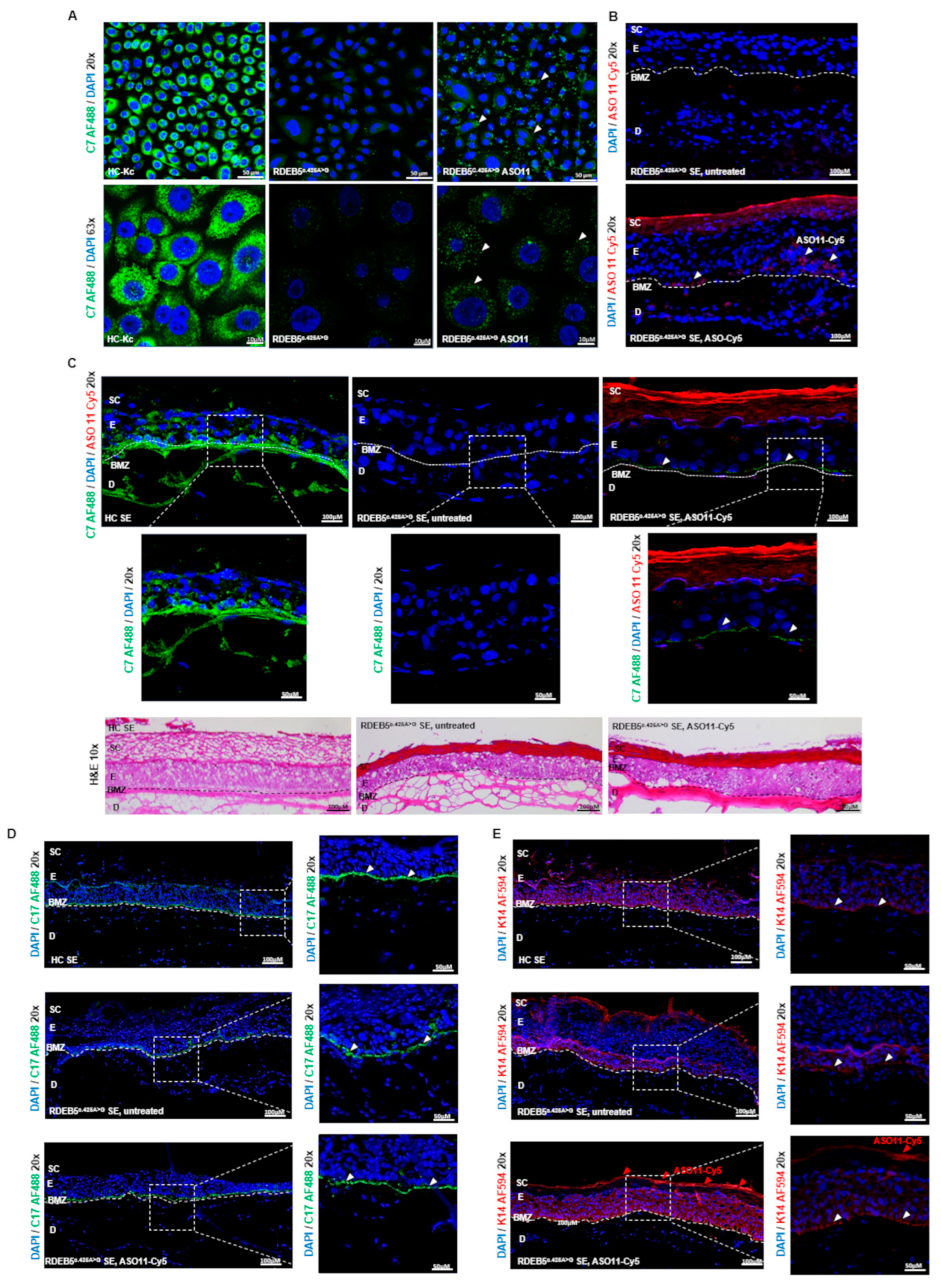
2. Results
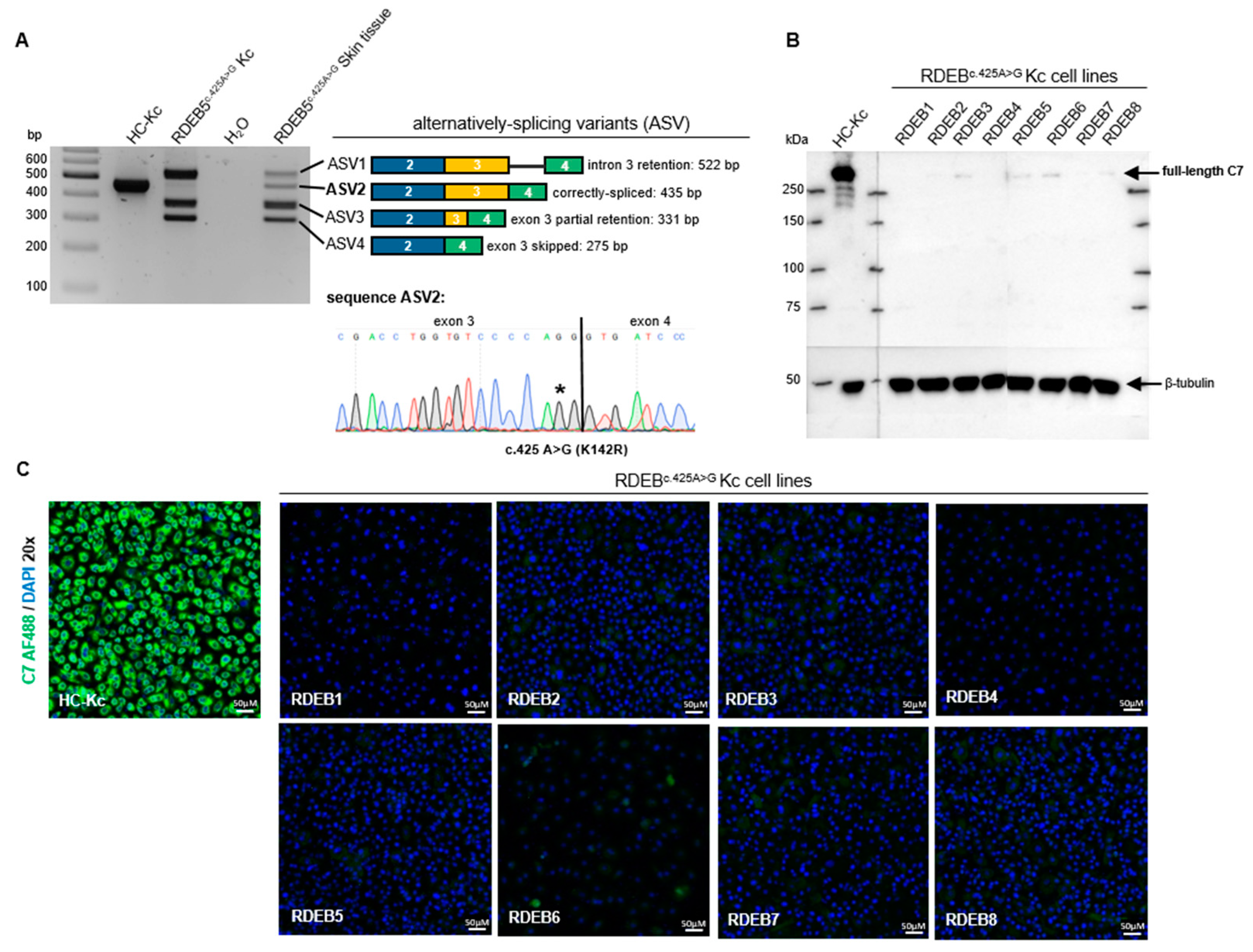
2.1. Characterization of the Splicing Pattern in RDEB Keratinocytes
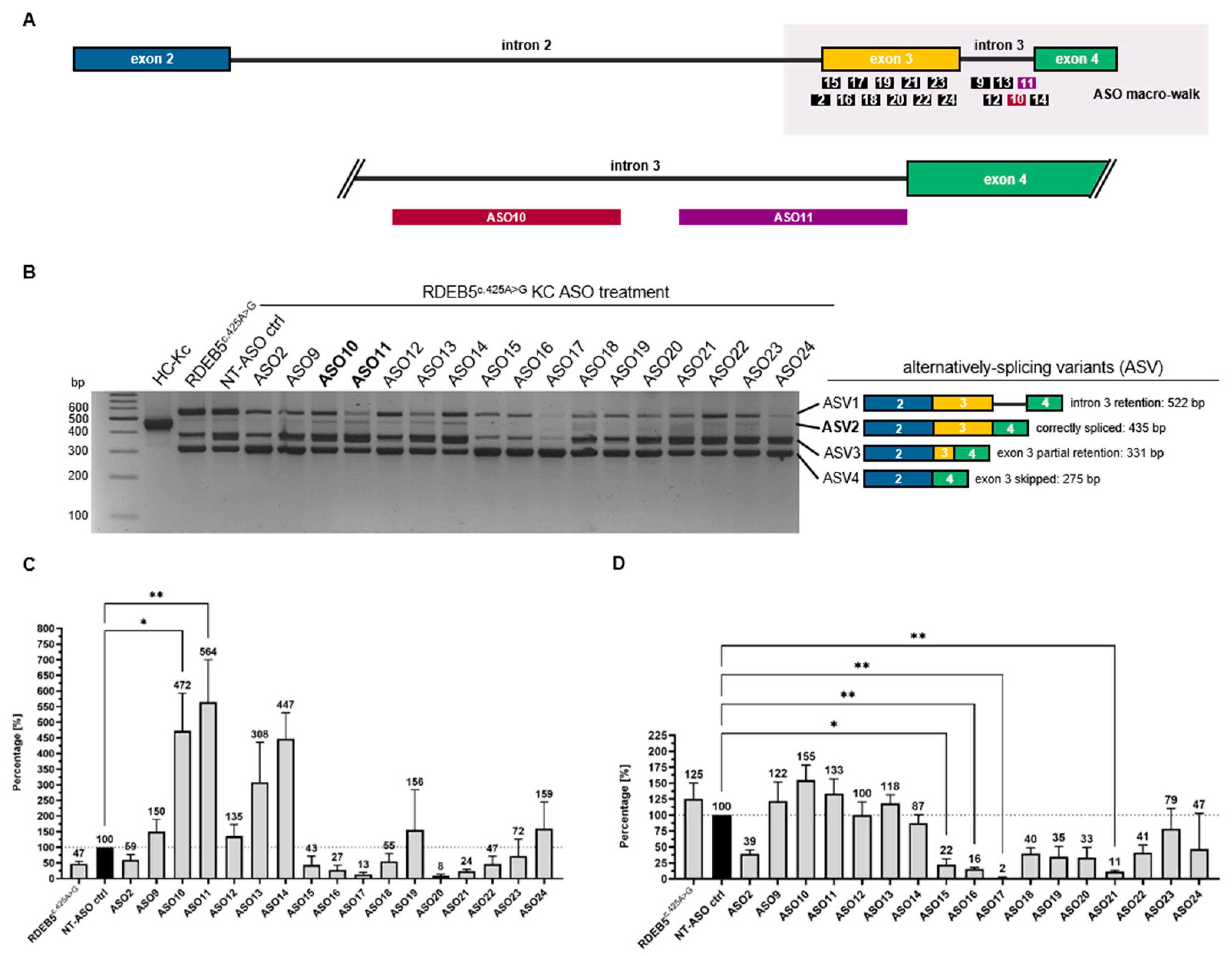
2.2. Initial Screening Identifies ASOs Capable of Modulating Splicing in RDEB Keratinocytes
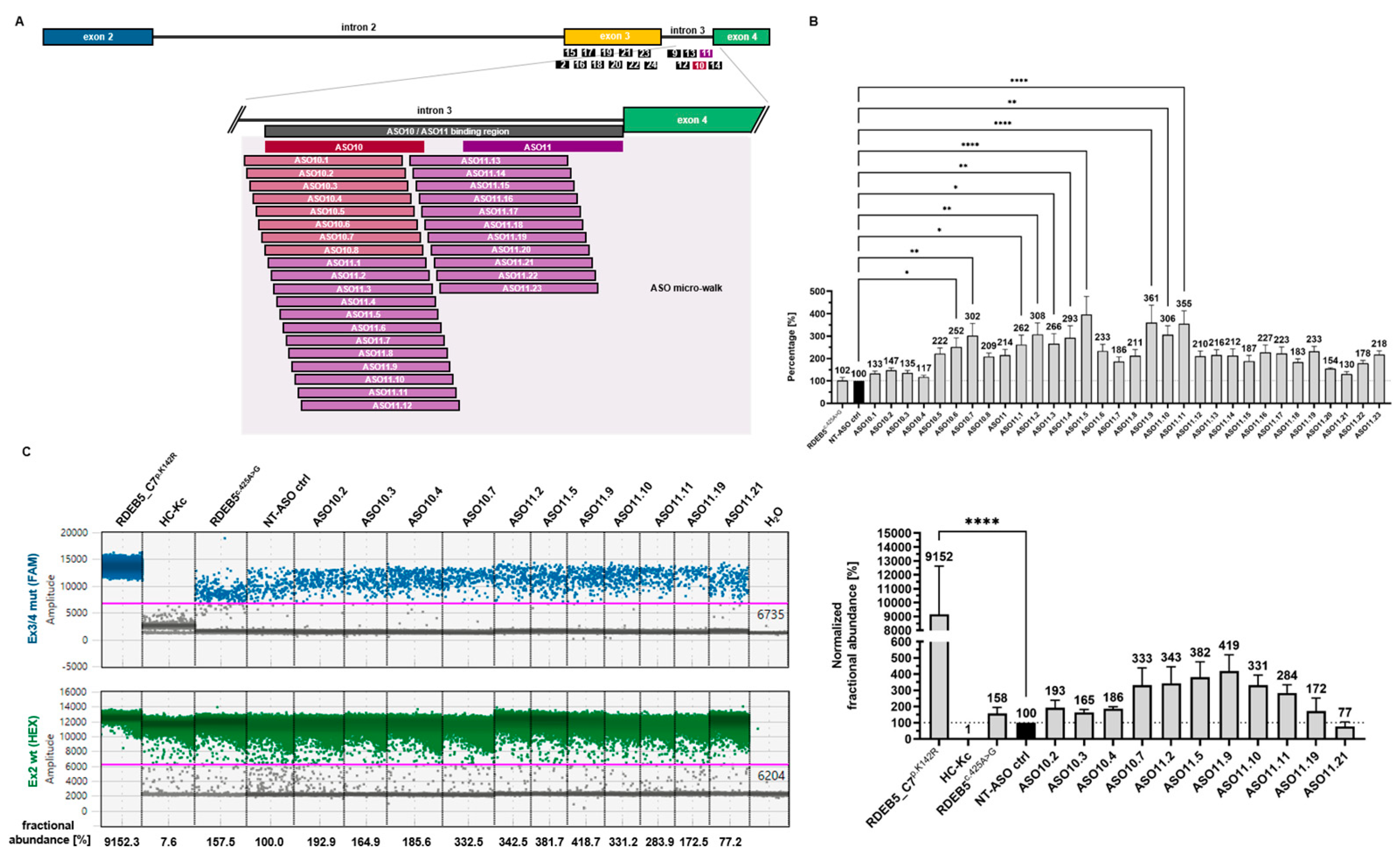
2.3. ASO Micro-Walk along the ASO10/ASO11 Binding Region
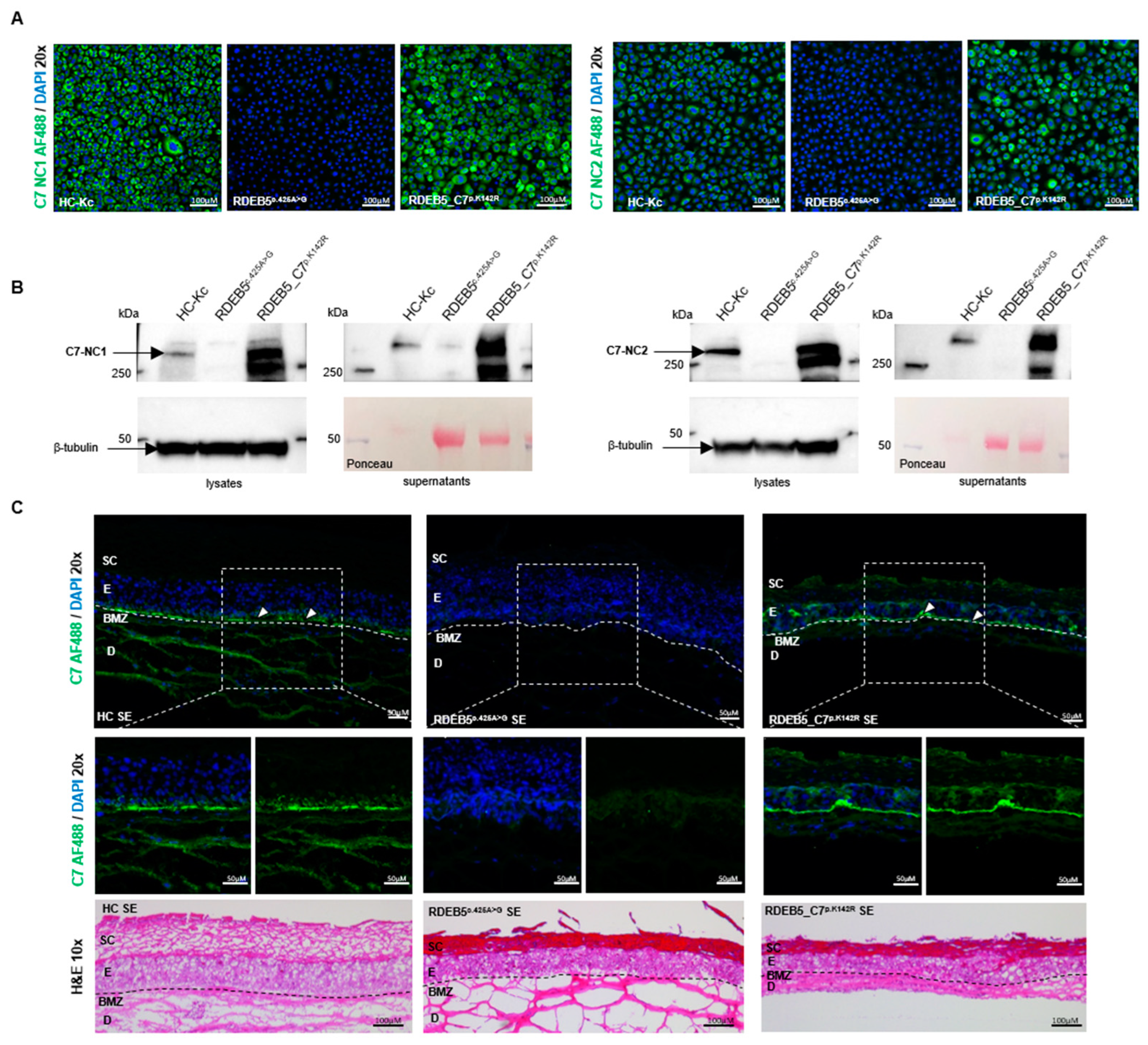
2.4. Impact of ASO Treatment on C7 Protein Expression
3. Discussion
4. Materials and Methods
4.1. Cell Line Generation
4.2. ASO Synthesis
4.3. Constructs
4.4. Transfection
4.5. RNA Isolation
4.6. RT-PCR
4.7. sqRT-PCR
4.8. Immunofluorescence Staining of C7 in Keratinocytes and Fibroblasts
4.9. Densitometric Analysis of Splice Products via Fiji (ImageJ)
4.10. Protein Isolation and Western Blot Analysis
4.11. Generation of DDC642 Liposomes
4.12. Generation and Treatment of Skin Equivalents
4.13. Immunofluorescence Staining of Skin Equivalents
4.14. Immunohistochemistry Staining of Skin Equivalents
4.15. ddPCR
4.16. TECAN Spark® Measurement
4.17. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K. RNA therapy: Rich history, various applications and unlimited future prospects. Exp. Mol. Med. 2022, 54, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Maire, S.; Gaillard, M.C.; Sahel, J.A.; Hantraye, P.; Bemelmans, A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, L.; Wang, X.; Jin, H. RNA-based therapeutics: An overview and prospectus. Cell Death Dis. 2022, 13, 644. [Google Scholar] [CrossRef] [PubMed]
- Hainzl, S.; Peking, P.; Kocher, T.; Murauer, E.M.; Larcher, F.; Del Rio, M.; Duarte, B.; Steiner, M.; Klausegger, A.; Bauer, J.W.; et al. COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2017, 25, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Hua, Y.; Krainer, A.R. Antisense-mediated exon inclusion. Methods Mol. Biol. 2012, 867, 307–323. [Google Scholar] [CrossRef]
- Sazani, P.; Gemignani, F.; Kang, S.H.; Maier, M.A.; Manoharan, M.; Persmark, M.; Bortner, D.; Kole, R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat. Biotechnol. 2002, 20, 1228–1233. [Google Scholar] [CrossRef]
- Sogaard, P.P.; Lind, M.; Christiansen, C.R.; Petersson, K.; Clauss, A.; Caffarel-Salvador, E. Future Perspectives of Oral Delivery of Next Generation Therapies for Treatment of Skin Diseases. Pharmaceutics 2021, 13, 1722. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Nystrom, A.; Saeidian, A.H.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol. 2018, 71, 313–329. [Google Scholar] [CrossRef]
- Nystrom, A.; Bruckner-Tuderman, L.; Kiritsi, D. Dystrophic Epidermolysis Bullosa: Secondary Disease Mechanisms and Disease Modifiers. Front. Genet. 2021, 12, 737272. [Google Scholar] [CrossRef] [PubMed]
- Csikos, M.; Szocs, H.I.; Laszik, A.; Mecklenbeck, S.; Horvath, A.; Karpati, S.; Bruckner-Tuderman, L. High frequency of the 425A→G splice-site mutation and novel mutations of the COL7A1 gene in central Europe: Significance for future mutation detection strategies in dystrophic epidermolysis bullosa. Br. J. Dermatol. 2005, 152, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Gardella, R.; Belletti, L.; Zoppi, N.; Marini, D.; Barlati, S.; Colombi, M. Identification of two splicing mutations in the collagen type VII gene (COL7A1) of a patient affected by the localisata variant of recessive dystrophic epidermolysis bullosa. Am. J. Hum. Genet. 1996, 59, 292–300. [Google Scholar] [PubMed]
- FDA. FDA Approves First Drug for Spinal Muscular Atrophy. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy (accessed on 18 November 2023).
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Lim, S.R.; Hertel, K.J. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J. Biol. Chem. 2001, 276, 45476–45483. [Google Scholar] [CrossRef]
- Breuel, S.; Vorm, M.; Brauer, A.U.; Owczarek-Lipska, M.; Neidhardt, J. Combining Engineered U1 snRNA and Antisense Oligonucleotides to Improve the Treatment of a BBS1 Splice Site Mutation. Mol. Ther. Nucleic Acids 2019, 18, 123–130. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Hollinger, K.; Bhattacharya, D.; Singh, R.N. An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA 2010, 16, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Desmet, E.; Bracke, S.; Forier, K.; Taevernier, L.; Stuart, M.C.; De Spiegeleer, B.; Raemdonck, K.; Van Gele, M.; Lambert, J. An elastic liposomal formulation for RNAi-based topical treatment of skin disorders: Proof-of-concept in the treatment of psoriasis. Int. J. Pharm. 2016, 500, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ablinger, M.; Lettner, T.; Friedl, N.; Potocki, H.; Palmetzhofer, T.; Koller, U.; Illmer, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; et al. Personalized Development of Antisense Oligonucleotides for Exon Skipping Restores Type XVII Collagen Expression in Junctional Epidermolysis Bullosa. Int. J. Mol. Sci. 2021, 22, 3326. [Google Scholar] [CrossRef]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef]
- De Rosa, L.; Carulli, S.; Cocchiarella, F.; Quaglino, D.; Enzo, E.; Franchini, E.; Giannetti, A.; De Santis, G.; Recchia, A.; Pellegrini, G.; et al. Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Rep. 2014, 2, 1–8. [Google Scholar] [CrossRef]
- Gurevich, I.; Agarwal, P.; Zhang, P.; Dolorito, J.A.; Oliver, S.; Liu, H.; Reitze, N.; Sarma, N.; Bagci, I.S.; Sridhar, K.; et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: A phase 1 and 2 trial. Nat. Med. 2022, 28, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Guide, S.V.; Gonzalez, M.E.; Bagci, I.S.; Agostini, B.; Chen, H.; Feeney, G.; Steimer, M.; Kapadia, B.; Sridhar, K.; Quesada Sanchez, L.; et al. Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 2022, 387, 2211–2219. [Google Scholar] [CrossRef]
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Investig. Dermatol. 2019, 139, 1221–1226. [Google Scholar] [CrossRef]
- Welponer, T.; Prodinger, C.; Pinon-Hofbauer, J.; Hintersteininger, A.; Breitenbach-Koller, H.; Bauer, J.W.; Laimer, M. Clinical Perspectives of Gene-Targeted Therapies for Epidermolysis Bullosa. Dermatol. Ther. 2021, 11, 1175–1197. [Google Scholar] [CrossRef]
- FDA. VYJUVEK. Available online: https://www.fda.gov/vaccines-blood-biologics/vyjuvek (accessed on 27 November 2023).
- Matsuo, M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021, 4, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef] [PubMed]
- Bornert, O.; Hogervorst, M.; Nauroy, P.; Bischof, J.; Swildens, J.; Athanasiou, I.; Tufa, S.F.; Keene, D.R.; Kiritsi, D.; Hainzl, S.; et al. QR-313, an Antisense Oligonucleotide, Shows Therapeutic Efficacy for Treatment of Dominant and Recessive Dystrophic Epidermolysis Bullosa: A Preclinical Study. J. Investig. Dermatol. 2021, 141, 883–893.e6. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Bornert, O.; Nystrom, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; van den Akker, P.C.; Pasmooij, A.M. Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Decha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003, 10, 120–125. [Google Scholar] [CrossRef]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef]
- Craig, K.; Abrams, M.; Amiji, M. Recent preclinical and clinical advances in oligonucleotide conjugates. Expert Opin. Drug Deliv. 2018, 15, 629–640. [Google Scholar] [CrossRef]
- Huang, S.; Hao, X.Y.; Li, Y.J.; Wu, J.Y.; Xiang, D.X.; Luo, S. Nonviral delivery systems for antisense oligonucleotide therapeutics. Biomater. Res. 2022, 26, 49. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, P.; Ceccoli, J. Advances in the Application and Impact of MicroRNAs as Therapies for Skin Disease. BioDrugs 2017, 31, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Nichols, J.G.; De Hoyos, C.L.; Sun, H.; Zhang, L.; Crooke, S.T. Golgi-58K can re-localize to late endosomes upon cellular uptake of PS-ASOs and facilitates endosomal release of ASOs. Nucleic Acids Res. 2021, 49, 8277–8293. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.C.; Del Agua, N.; Lwin, S.M.; Hsu, C.K.; McGrath, J.A. Innovations in the Treatment of Dystrophic Epidermolysis Bullosa (DEB): Current Landscape and Prospects. Ther. Clin. Risk Manag. 2023, 19, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Kumbhojkar, N.; Reilly, C.; Dharamdasani, V.; Ukidve, A.; Ingber, D.E.; Mitragotri, S. Treatment of psoriasis with NFKBIZ siRNA using topical ionic liquid formulations. Sci. Adv. 2020, 6, eabb6049. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell Biol. 2004, 24, 10505–10514. [Google Scholar] [CrossRef]
- Ong, A.A.L.; Tan, J.; Bhadra, M.; Dezanet, C.; Patil, K.M.; Chong, M.S.; Kierzek, R.; Decout, J.L.; Roca, X.; Chen, G. RNA Secondary Structure-Based Design of Antisense Peptide Nucleic Acids for Modulating Disease-Associated Aberrant Tau Pre-mRNA Alternative Splicing. Molecules 2019, 24, 3020. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid. Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Liang, X.H.; Crooke, R.M.; Baker, B.F.; Geary, R.S. Antisense drug discovery and development technology considered in a pharmacological context. Biochem. Pharmacol. 2021, 189, 114196. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Bornert, O.; Kocher, T.; Gretzmeier, C.; Liemberger, B.; Hainzl, S.; Koller, U.; Nystrom, A. Generation of rabbit polyclonal human and murine collagen VII monospecific antibodies: A useful tool for dystrophic epidermolysis bullosa therapy studies. Matrix Biol. Plus 2019, 4, 100017. [Google Scholar] [CrossRef]
- Kocher, T.; March, O.P.; Bischof, J.; Liemberger, B.; Hainzl, S.; Klausegger, A.; Hoog, A.; Strunk, D.; Bauer, J.W.; Koller, U. Predictable CRISPR/Cas9-Mediated COL7A1 Reframing for Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2020, 140, 1985–1993.e5. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Mutation | Exon | Cell Type |
|---|---|---|---|
| RDEB1 | c.425A > G/c.6841G > T | exon 3, exon 63 | keratinocytes |
| RDEB2 | c.425A > G/c.520G > A | exon 3, exon 4 | keratinocytes |
| RDEB3 | c.425A > G/c.520G > A | exon 3, exon 4 | keratinocytes |
| RDEB4 | c.425A > G/c.4027C > T | exon 3, exon 34 | keratinocytes |
| RDEB5 | c.425A > G/c.425A > G | exon 3, exon 3 | keratinocytes |
| RDEB5 | c.425A > G/c.425A > G | exon 3, exon 3 | fibroblasts |
| RDEB6 | c.425A > G/c.682 + 5G > A | exon 3, intron 5 | keratinocytes |
| RDEB7 | c.425A > G/c.425A > G | exon 3, exon 3 | keratinocytes |
| RDEB8 | c.425A > G/c.425A > G | exon 3, exon 3 | keratinocytes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hainzl, S.; Trattner, L.; Liemberger, B.; Bischof, J.; Kocher, T.; Ablinger, M.; Nyström, A.; Obermayer, A.; Klausegger, A.; Guttmann-Gruber, C.; et al. Splicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa. Int. J. Mol. Sci. 2024, 25, 761. https://doi.org/10.3390/ijms25020761
Hainzl S, Trattner L, Liemberger B, Bischof J, Kocher T, Ablinger M, Nyström A, Obermayer A, Klausegger A, Guttmann-Gruber C, et al. Splicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa. International Journal of Molecular Sciences. 2024; 25(2):761. https://doi.org/10.3390/ijms25020761
Chicago/Turabian StyleHainzl, Stefan, Lisa Trattner, Bernadette Liemberger, Johannes Bischof, Thomas Kocher, Michael Ablinger, Alexander Nyström, Astrid Obermayer, Alfred Klausegger, Christina Guttmann-Gruber, and et al. 2024. "Splicing Modulation via Antisense Oligonucleotides in Recessive Dystrophic Epidermolysis Bullosa" International Journal of Molecular Sciences 25, no. 2: 761. https://doi.org/10.3390/ijms25020761