
Genome-Wide Association Study for Maize Hybrid Performance in a Typical Breeder Population

 and
and
Abstract
:1. Introduction
2. Results
2.1. Genetic Characteristics of the Hybrid Population
2.2. GWAS Model Comparison in the Hybrid Population
2.3. GWAS Results Based on the KinADE Model
2.4. Allele Frequencies of QTNs and Favorable Genotype Accumulation Effects for the 10 Traits
2.5. Candidate Gene Identification
3. Discussion
3.1. The Main Genetic Effects of Different Traits with Existential Discrepancy
3.2. Advantages of Genetic Analysis in Hybrid Populations
3.3. The Genetic Distance of Core Tester Lines within the Heterotic Group Influences Little to GWAS Using the Inter-Heterotic-Group Hybrid Population
3.4. The Inter-Heterotic-Group Hybrid Population Can Be Optimized by Increasing the Number of Core Tester Lines without Increasing Resource Cost
3.5. Heterozygosity Contributes Significantly to Hybrid Yield
3.6. Using Genomically Predicted Phenotypes Does Not Increase the Power of the GWAS
3.7. Current Research on Candidate Genes
3.8. Predominant Alleles among Populations
4. Materials and Methods
4.1. Materials and Field Experiment Design
4.2. Genetic Characteristics Analysis
4.3. Genome-Wide Association Studies in Hybrid Populations
4.4. Candidate Gene Selection and Annotation of Associated SNPs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mackay, T.F. A-maize-ing diversity. Science 2009, 325, 688. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Cooper, M.; Gogerty, J.; Löffler, C.; Borcherding, D.; Wright, K. Maize. In Yield Gains in Major US Field Crops; Smith, S., Diers, B., Specht, J., Carver, B., Eds.; CSSA Special Publications: Madison, WI, USA, 2014; Volume 33, pp. 125–171. [Google Scholar]
- Duvick, D.N.; Smith, J.S.C.; Cooper, M. Long-term Selection in a Commercial Hybrid Maize Breeding Program. In Plant Breeding Reviews; Janick, J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 109–152. [Google Scholar]
- Reif, J.C.; Hamrit, S.; Heckenberger, M.; Schipprack, W.; Maurer, H.P.; Bohn, M.; Melchinger, A.E. Trends in genetic diversity among European maize cultivars and their parental components during the past 50 years. Theor. Appl. Genet. 2005, 111, 838. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Gaut, B.S.; McMullen, M.D. Molecular and functional diversity of maize. Curr. Opin. Plant Biol. 2006, 9, 172. [Google Scholar] [CrossRef]
- Jiao, Y.P.; Peluso, P.; Shi, J.H.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.H.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524. [Google Scholar] [CrossRef]
- Sun, S.L.; Zhou, Y.S.; Chen, J.; Shi, J.P.; Zhao, H.M.; Zhao, H.N.; Song, W.B.; Zhang, M.; Cui, Y.; Dong, X.M.; et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289. [Google Scholar] [CrossRef]
- Li, C.H.; Song, W.; Luo, Y.F.; Gao, S.H.; Zhang, R.Y.; Shi, Z.; Wang, X.Q.; Wang, R.H.; Wang, F.G.; Wang, J.D.; et al. The HuangZaoSi Maize Genome Provides Insights into Genomic Variation and Improvement History of Maize. Mol. Plant 2019, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.; Ersoz, E.; Flint-Garcia, S.; Garcia, A.; Glaubitz, J.C.; et al. The genetic architecture of maize flowering time. Science 2009, 325, 714. [Google Scholar] [CrossRef] [PubMed]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; McMullen, M.D.; Ware, D.; et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163. [Google Scholar]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159. [Google Scholar] [CrossRef]
- Liu, J.; Seetharam, A.S.; Chougule, K.; Ou, S.; Swentowsky, K.W.; Gent, J.I.; Llaca, V.; Woodhouse, M.R.; Manchanda, N.; Presting, G.G.; et al. Gapless assembly of maize chromosomes using long-read technologies. Genome Biol. 2020, 21, 121. [Google Scholar] [CrossRef]
- Liu, S.X.; Wang, X.L.; Wang, H.W.; Xin, H.B.; Yang, X.H.; Yan, J.B.; Li, J.S.; Tran, L.S.P.; Shinozaki, K.; Yamaguchi-Shinozaki, K.; et al. Genome-Wide Analysis of ZmDREB Genes and Their Association with Natural Variation in Drought Tolerance at Seedling Stage of Zea mays L. PLoS Genet. 2013, 9, e1003790. [Google Scholar]
- Wen, W.; Li, D.; Li, X.; Gao, Y.; Li, W.; Li, H.; Liu, J.; Liu, H.; Chen, W.; Luo, J.; et al. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014, 5, 3438. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Han, Y.J.; Liu, H.J.; Wang, X.F.; Sun, J.M.; Zhao, B.H.; Li, W.Y.; Tian, J.G.; Liang, Y.M.; Yan, J.B.; et al. Genome-Wide Association Analyses Reveal the Importance of Alternative Splicing in Diversifying Gene Function and Regulating Phenotypic Variation in Maize. Plant Cell 2018, 30, 1404. [Google Scholar] [CrossRef]
- Li, W.Q.; Yu, Y.H.; Wang, L.X.; Luo, Y.; Peng, Y.; Xu, Y.C.; Liu, X.G.; Wu, S.S.; Jian, L.M.; Xu, J.T.; et al. The genetic architecture of the dynamic changes in grain moisture in maize. Plant Biotechnol. J. 2021, 19, 1195. [Google Scholar] [CrossRef]
- Li, D.; Li, G.; Wang, H.; Guo, Y.; Wang, M.; Lu, X.; Luo, Z.; Zhu, X.; Weiss, T.M.; Roller, S.; et al. Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize. Int. J. Mol. Sci. 2022, 23, 13943. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, D.; Xu, Z.; Gu, R.; Wang, P.; Fu, J.; Wang, J.; Du, W.; Zhang, H. Dissection of the Genetic Basis of Yield Traits in Line per se and Testcross Populations and Identification of Candidate Genes for Hybrid Performance in Maize. Int. J. Mol. Sci. 2022, 23, 5074. [Google Scholar] [CrossRef]
- Zeng, T.; Meng, Z.; Yue, R.; Lu, S.; Li, W.; Li, W.; Meng, H.; Sun, Q. Genome wide association analysis for yield related traits in maize. BMC Plant Biol. 2022, 22, 449. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, C.; Liu, X.G.; Guo, Z.F.; Xu, X.J.; Wang, S.H.; Xie, C.X.; Li, W.X.; Zou, C.; Xu, Y.B. Development of a multiple-hybrid population for genome-wide association studies: Theoretical consideration and genetic mapping of flowering traits in maize. Sci. Rep. 2017, 7, 40239. [Google Scholar] [PubMed]
- Zhao, Y.; Wang, H.S.; Bo, C.; Dai, W.; Zhang, X.G.; Cai, R.H.; Gu, L.J.; Ma, Q.; Jiang, H.Y.; Zhu, J.; et al. Genome-wide association study of maize plant architecture using F-1 populations. Plant Mol. Biol. 2019, 99, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wan, J.; He, L.; Lan, H.; Li, L. Genome-Wide Association Analysis of Plant Height Using the Maize F1 Population. Plants 2019, 8, 432. [Google Scholar] [PubMed]
- Liu, H.; Wang, Q.; Chen, M.; Ding, Y.; Yang, X.; Liu, J.; Li, X.; Zhou, C.; Tian, Q.; Lu, Y.; et al. Genome-wide identification and analysis of heterotic loci in three maize hybrids. Plant Biotechnol. J. 2020, 18, 185. [Google Scholar]
- Xiao, Y.; Jiang, S.; Cheng, Q.; Wang, X.; Yan, J.; Zhang, R.; Qiao, F.; Ma, C.; Luo, J.; Li, W.; et al. The genetic mechanism of heterosis utilization in maize improvement. Genome Biol. 2021, 22, 148. [Google Scholar]
- Huang, X.H.; Yang, S.H.; Gong, J.Y.; Zhao, Y.; Feng, Q.; Gong, H.; Li, W.J.; Zhan, Q.L.; Cheng, B.Y.; Xia, J.H.; et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun. 2015, 6, 6258. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Schmidt, R.H.; Zhao, Y.S.; Reif, J.C. A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat. Genet. 2017, 49, 1741. [Google Scholar] [CrossRef]
- Dudley, J.W.; Johnson, G.R.J.C.S. Epistatic Models Improve Prediction of Performance in Corn. Crop Sci. 2009, 49, 763. [Google Scholar]
- Kusmec, A.; Srinivasan, S.; Nettleton, D.; Schnable, P.S. Distinct genetic architectures for phenotype means and plasticities in Zea mays. Nat. Plants 2017, 3, 715. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, X.; Li, Y.; Shi, Y.; Song, Y.; Zhang, D.; Li, Y.; Wang, T. Genetic architecture of phenotypic means and plasticities of kernel size and weight in maize. Theor. Appl. Genet. 2019, 132, 3309. [Google Scholar] [CrossRef]
- Li, G.L.; Dong, Y.; Zhao, Y.S.; Tian, X.K.; Wurschum, T.; Xue, J.Q.; Chen, S.J.; Reif, J.C.; Xu, S.T.; Liu, W.X. Genome-wide prediction in a hybrid maize population adapted to Northwest China. Crop J. 2020, 8, 830. [Google Scholar]
- Yang, C.; Zhang, L.; Jia, A.; Rong, T. Identification of QTL for maize grain yield and kernel-related traits. J. Genet. 2016, 95, 239. [Google Scholar] [CrossRef]
- Liu, J.; Yu, H.; Liu, Y.; Deng, S.; Liu, Q.; Liu, B.; Xu, M. Genetic dissection of grain water content and dehydration rate related to mechanical harvest in maize. BMC Plant Biol. 2020, 20, 118. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.Q.; Tang, B.J.; Ding, Y.; Xia, L.K.; Qi, J.S.; Mu, X.Y.; Gu, L.M.; Lu, D.W.; Chen, Y.H. Molecular mapping of quantitative trait loci for grain moisture at harvest and field grain drying rate in maize (Zea mays L.). Physiol. Plant. 2020, 169, 64. [Google Scholar] [PubMed]
- Flint-Garcia, S.A.; Jampatong, C.; Darrah, L.L.; McMullen, M.D. Quantitative trait locus analysis of stalk strength in four maize populations. Crop Sci. 2003, 43, 13. [Google Scholar] [CrossRef]
- Liu, X.G.; Hu, X.J.; Li, K.; Liu, Z.F.; Wu, Y.J.; Wang, H.W.; Huang, C.L. Genetic mapping and genomic selection for maize stalk strength. Bmc Plant Biol. 2020, 20, 196. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, S.H.; Mock, J.J. Genetics of tassel branch number in maize and its implications for a selection program for small tassel size. Theor. Appl. Genet. 1978, 53, 265. [Google Scholar]
- Tanaka, W.; Pautler, M.; Jackson, D.; Hirano, H.Y. Grass meristems II: Inflorescence architecture, flower development and meristem fate. Plant Cell Physiol. 2013, 54, 313. [Google Scholar] [CrossRef]
- Hoecker, N.; Keller, B.; Muthreich, N.; Chollet, D.; Descombes, P.; Piepho, H.P.; Hochholdinger, F. Comparison of maize (Zea mays L.) F1-hybrid and parental inbred line primary root transcriptomes suggests organ-specific patterns of nonadditive gene expression and conserved expression trends. Genetics 2008, 179, 1275. [Google Scholar] [CrossRef]
- Krieger, U.; Lippman, Z.B.; Zamir, D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 2010, 42, 459. [Google Scholar] [CrossRef]
- Yang, J.L.; Mezmouk, S.; Baumgarten, A.; Buckler, E.S.; Guill, K.E.; McMullen, M.D.; Mumm, R.H.; Ross-Ibarra, J. Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize. PLoS Genet. 2017, 13, e1007019. [Google Scholar] [CrossRef]
- Zhou, G.; Chen, Y.; Yao, W.; Zhang, C.; Xie, W.; Hua, J.; Xing, Y.; Xiao, J.; Zhang, Q. Genetic composition of yield heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 2012, 109, 15847. [Google Scholar] [CrossRef]
- Luo, J.H.; Wang, M.; Jia, G.F.; He, Y. Transcriptome-wide analysis of epitranscriptome and translational efficiency associated with heterosis in maize. J. Exp. Bot. 2021, 72, 2933. [Google Scholar] [PubMed]
- Hu, X.; Wang, H.; Diao, X.; Liu, Z.; Li, K.; Wu, Y.; Liang, Q.; Wang, H.; Huang, C. Transcriptome profiling and comparison of maize ear heterosis during the spikelet and floret differentiation stages. BMC Genom. 2016, 17, 959. [Google Scholar] [CrossRef]
- Huang, Y.; Qi, Z.; Li, J.; You, J.; Zhang, X.; Wang, M. Genetic interrogation of phenotypic plasticity informs genome-enabled breeding in cotton. J. Genet. Genom. 2023, 50, 971–982. [Google Scholar] [CrossRef]
- Liu, L.; Du, Y.; Huo, D.; Wang, M.; Shen, X.; Yue, B.; Qiu, F.; Zheng, Y.; Yan, J.; Zhang, Z. Genetic architecture of maize kernel row number and whole genome prediction. Theor. Appl. Genet. 2015, 128, 2243. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Dey, N.; Kim, C.S.; Bharti, A.K.; Rudd, S.; Mayer, K.F.; Larkins, B.A.; Becraft, P.; Messing, J. Characterization of the maize endosperm transcriptome and its comparison to the rice genome. Genome Res. 2004, 14, 1932. [Google Scholar] [PubMed]
- Tappiban, P.; Ying, Y.; Xu, F.; Bao, J. Proteomics and Post-Translational Modifications of Starch Biosynthesis-Related Proteins in Developing Seeds of Rice. Int. J. Mol. Sci. 2021, 22, 5901. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Li, Y.; Zhang, Q.; Liu, Q.; Zhai, H.; Zhao, N.; Yang, Y.; He, S. Plastidial Phosphoglucomutase (pPGM) Overexpression Increases the Starch Content of Transgenic Sweet Potato Storage Roots. Genes 2022, 13, 2234. [Google Scholar] [CrossRef] [PubMed]
- Monroe-Augustus, M.; Zolman, B.K.; Bartel, B. IBR5, a dual-specificity phosphatase-like protein modulating auxin and abscisic acid responsiveness in Arabidopsis. Plant Cell 2003, 15, 2979. [Google Scholar]
- Mayank, P.; Grossman, J.; Wuest, S.; Boisson-Dernier, A.; Roschitzki, B.; Nanni, P.; Nuhse, T.; Grossniklaus, U. Characterization of the phosphoproteome of mature Arabidopsis pollen. Plant J. 2012, 72, 89. [Google Scholar] [CrossRef]
- Wu, J.; Chen, L.; Chen, M.; Zhou, W.; Dong, Q.; Jiang, H.; Cheng, B. The DOF-Domain Transcription Factor ZmDOF36 Positively Regulates Starch Synthesis in Transgenic Maize. Front. Plant Sci. 2019, 10, 465. [Google Scholar] [CrossRef]
- Yan, J.; Wu, Y.; Li, W.; Qin, X.; Wang, Y.; Yue, B. Genetic mapping with testcrossing associations and F2:3 populations reveals the importance of heterosis in chilling tolerance at maize seedling stage. Sci. Rep. 2017, 7, 3232. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.; Li, Y.; Yan, W.; Chang, Z.; Ni, H.; Chen, Z.; Wu, J.; Xu, C.; Deng, X.W.; et al. GDSL esterase/lipases OsGELP34 and OsGELP110/OsGELP115 are essential for rice pollen development. J. Integr. Plant Biol. 2020, 62, 1574. [Google Scholar]
- Zhao, H.; Ma, B.; Duan, K.X.; Li, X.K.; Lu, X.; Yin, C.C.; Tao, J.J.; Wei, W.; Zhang, W.K.; Xin, P.Y.; et al. The GDSL Lipase MHZ11 Modulates Ethylene Signaling in Rice Roots. Plant Cell 2020, 32, 1626. [Google Scholar] [CrossRef]
- Dai, Y.; Sun, X.; Wang, C.; Li, F.; Zhang, S.; Zhang, H.; Li, G.; Yuan, L.; Chen, G.; Sun, R.; et al. Gene co-expression network analysis reveals key pathways and hub genes in Chinese cabbage (Brassica rapa L.) during vernalization. BMC Genom. 2021, 22, 236. [Google Scholar] [CrossRef]
- Sun, L.; Miao, Z.; Cai, C.; Zhang, D.; Zhao, M.; Wu, Y.; Zhang, X.; Swarm, S.A.; Zhou, L.; Zhang, Z.J.; et al. GmHs1-1, encoding a calcineurin-like protein, controls hard-seededness in soybean. Nat. Genet. 2015, 47, 939. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, K.; Xu, C.; Zou, C.; Xie, C.; Xu, Y.; Li, W.X. Strand-specific RNA-Seq transcriptome analysis of genotypes with and without low-phosphorus tolerance provides novel insights into phosphorus-use efficiency in maize. BMC Plant Biol. 2016, 16, 222. [Google Scholar] [CrossRef]
- Rosa, M.; Abraham-Juarez, M.J.; Lewis, M.W.; Fonseca, J.P.; Tian, W.; Ramirez, V.; Luan, S.; Pauly, M.; Hake, S. The Maize Mid-Complementing Activity Homolog Cell Number Regulator13/Narrow Odd Dwarf Coordinates Organ Growth and Tissue Patterning. Plant Cell 2017, 29, 474. [Google Scholar] [PubMed]
- Peiffer, J.A.; Flint-Garcia, S.A.; De Leon, N.; McMullen, M.D.; Kaeppler, S.M.; Buckler, E.S. The genetic architecture of maize stalk strength. PLoS ONE 2013, 8, e67066. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Romay, M.C.; Gore, M.A.; Flint-Garcia, S.A.; Zhang, Z.; Millard, M.J.; Gardner, C.A.; McMullen, M.D.; Holland, J.B.; Bradbury, P.J.; et al. The genetic architecture of maize height. Genetics 2014, 196, 1337. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Shi, Y.; Song, Y.; Zhang, D.; Li, C.; Buckler, E.S.; Li, Y.; Zhang, Z.; Wang, T. Joint-linkage mapping and GWAS reveal extensive genetic loci that regulate male inflorescence size in maize. Plant Biotechnol. J. 2016, 14, 1551. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Qu, J.; Wang, Y.; Chang, L.; He, K.; Guo, D.; Zhang, X.; Xu, S.; Xue, J. Genetic characterization of inbred lines from Shaan A and B groups for identifying loci associated with maize grain yield. BMC Genet. 2018, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406. [Google Scholar]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z. Mapping Quantitative Trait Loci by Controlling Polygenic Background Effects. Genetics 2013, 195, 1209. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Stamier, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Xu, S.; Gou, X.; Zhang, H.; Cheng, Q.; Wang, X.; Ma, C.; Xue, J. Time-resolved multiomics analysis of the genetic regulation of maize kernel moisture. Crop J. 2023, 11, 247. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
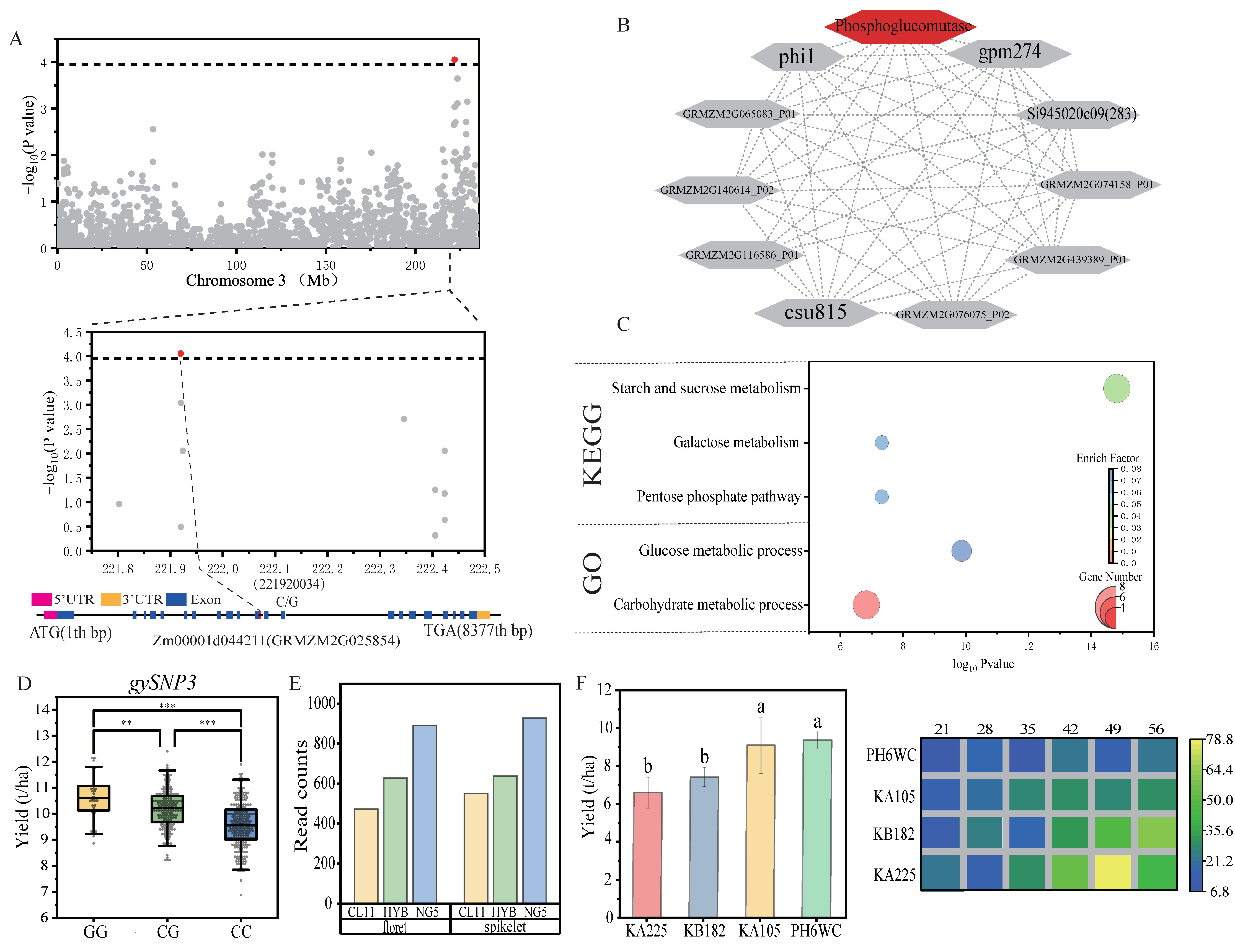{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | SNP | Chr | Position | Genotype | Mode * | Effect | −log10P | PVE (%) |
|---|---|---|---|---|---|---|---|---|
| GY | gySNP1 | 1 | 215245696 | T/A | Add | −0.5 | 4.0 | 6.7 |
| gySNP2 | 1 | 73916274 | G/A | Add/Dom | 0.92 | 5.3 | 2.1 | |
| gySNP3 | 3 | 221920034 | C/G | Add | −0.48 | 4.1 | 5.7 | |
| gySNP4 | 5 | 209076386 | G/T | Add | −0.56 | 4.5 | 12.6 | |
| Sum | 27.1 | |||||||
| GM | gmSNP1 | 2 | 1263421 | A/G | Add | 0.48 | 4.2 | 7.5 |
| gmSNP2 | 5 | 15548317 | C/T | Add | −0.91 | 4.6 | 2.4 | |
| gmSNP3 | 7 | 157333857 | C/T | Add | −0.77 | 4.5 | 12.6 | |
| gmSNP4 | 8 | 129615225 | G/A | Add | 0.58 | 4.0 | 18.7 | |
| gmSNP5 | 9 | 152684176 | G/A | Add | −0.51 | 4.1 | 1.6 | |
| Sum | 42.8 | |||||||
| PH | phSNP1 | 5 | 208463873 | C/G | Add | 9.69 | 4.7 | 9.6 |
| phSNP2 | 5 | 215535364 | A/G | Add | 8.39 | 4.0 | 13.2 | |
| phSNP3 | 6 | 124049465 | C/G | Add | 9.64 | 4.1 | 8.6 | |
| phSNP4 | 7 | 176596934 | A/G | Add | −11.57 | 4.2 | 2.3 | |
| phSNP5 | 8 | 128340728 | C/A | Add/Dom | 18.63 | 4.8 | 3.4 | |
| phSNP6 | 10 | 147469999 | C/T | Add/Dom | 10.77 | 4.2 | 10.6 | |
| Sum | 47.7 | |||||||
| EH | ehSNP1 | 3 | 291054 | G/A | Add | −5.03 | 4.1 | 14.2 |
| ehSNP2 | 6 | 132789720 | A/G | Add | −7.04 | 4.4 | 8.8 | |
| ehSNP3 | 7 | 180279662 | G/A | Add | −6.88 | 4.8 | 14.9 | |
| ehSNP4 | 9 | 71431208 | C/A | Add/Dom | 10.61 | 4.3 | 6.5 | |
| ehSNP5 | 9 | 79091347 | C/G | Add/Dom | −10.57 | 4.1 | 1.7 | |
| ehSNP6 | 9 | 99136257 | C/T | Add/Dom | −7.2 | 3.9 | 1.4 | |
| Sum | 47.5 | |||||||
| ELL | ellSNP1 | 1 | 254318451 | G/A | Dom | 2.04 | 4.0 | 2.2 |
| ellSNP2 | 6 | 118971410 | C/T | Add/Dom | 3.5 | 4.1 | 5.4 | |
| ellSNP3 | 8 | 128340728 | C/A | Add/Dom | 4.97 | 4.0 | 9.6 | |
| Sum | 17.2 | |||||||
| ELW | elwSNP1 | 2 | 21687575 | T/A | Dom | −0.2 | 4.1 | 1.3 |
| elwSNP2 | 3 | 159442050 | A/G | Add | 0.45 | 4.4 | 8.2 | |
| elwSNP3 | 3 | 229142753 | G/C | Add | −0.4 | 5.3 | 2.5 | |
| elwSNP4 | 5 | 196015158 | C/T | Add/Dom | 0.48 | 4.3 | 1.1 | |
| elwSNP5 | 5 | 212255011 | G/A | Add | −0.36 | 4.1 | 7.6 | |
| elwSNP6 | 6 | 167994782 | G/A | Add | −0.29 | 4.1 | 22.8 | |
| elwSNP7 | 10 | 107630915 | T/C | Add | 0.41 | 4.5 | 15.6 | |
| Sum | 59.1 | |||||||
| ELA | elaSNP1 | 2 | 195895775 | G/T | Add/Dom | 43.84 | 4.1 | 2.1 |
| elaSNP2 | 3 | 163190634 | C/T | Add | −20.65 | 3.9 | 9.5 | |
| elaSNP3 | 6 | 167994769 | T/C | Add | −29.02 | 6.4 | 20.1 | |
| elaSNP4 | 7 | 140317639 | G/A | Add | 26.56 | 4.0 | 10.6 | |
| elaSNP5 | 7 | 142451671 | G/A | Add | 23.51 | 4.1 | 1.1 | |
| elaSNP6 | 9 | 142662042 | C/G | Add | 23.81 | 4.0 | 3.6 | |
| Sum | 47.0 | |||||||
| RPR_TIAG | tiagSNP1 | 1 | 244211691 | T/A | Dom | 2.59 | 4.2 | 1.3 |
| tiagSNP2 | 6 | 168202087 | G/A | Add | 3.2 | 4.3 | 4.2 | |
| tiagSNP3 | 7 | 1403785 | G/T | Add | −1.86 | 4.0 | 6.5 | |
| Sum | 12.0 | |||||||
| RPR_IUE | iueSNP1 | 1 | 251572876 | A/T | Add/Dom | 1.72 | 4.0 | 3.6 |
| iueSNP2 | 6 | 157939881 | T/C | Dom | −1.7 | 4.9 | 7.6 | |
| iueSNP3 | 9 | 140917978 | T/C | Add/Dom | 2.49 | 4.2 | 8.1 | |
| Sum | 19.3 | |||||||
| TBN | tbnSNP1 | 2 | 45027887 | G/A | Add/Dom | 1.05 | 4.7 | 1.4 |
| tbnSNP2 | 2 | 51045430 | A/C | Add/Dom | 1.24 | 5.1 | 6.0 | |
| tbnSNP3 | 4 | 186665498 | G/A | Add/Dom | −1.06 | 5.4 | 3.1 | |
| tbnSNP4 | 4 | 189101304 | A/T | Add/Dom | −0.98 | 5.3 | 2.2 | |
| tbnSNP5 | 5 | 201203454 | C/T | Dom | 0.62 | 4.5 | 5.6 | |
| tbnSNP6 | 6 | 134143465 | A/T | Dom | −0.6 | 4.0 | 2.7 | |
| Sum | 21.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Li, G.; Zhang, X.; Feng, Z.; Li, T.; Li, Z.; Xu, S.; Xu, S.; Liu, W.; Xue, J. Genome-Wide Association Study for Maize Hybrid Performance in a Typical Breeder Population. Int. J. Mol. Sci. 2024, 25, 1190. https://doi.org/10.3390/ijms25021190
Dong Y, Li G, Zhang X, Feng Z, Li T, Li Z, Xu S, Xu S, Liu W, Xue J. Genome-Wide Association Study for Maize Hybrid Performance in a Typical Breeder Population. International Journal of Molecular Sciences. 2024; 25(2):1190. https://doi.org/10.3390/ijms25021190
Chicago/Turabian StyleDong, Yuan, Guoliang Li, Xinghua Zhang, Zhiqian Feng, Ting Li, Zhoushuai Li, Shizhong Xu, Shutu Xu, Wenxin Liu, and Jiquan Xue. 2024. "Genome-Wide Association Study for Maize Hybrid Performance in a Typical Breeder Population" International Journal of Molecular Sciences 25, no. 2: 1190. https://doi.org/10.3390/ijms25021190