
Vitamin B12 Deficiency and the Nervous System: Beyond Metabolic Decompensation—Comparing Biological Models and Gaining New Insights into Molecular and Cellular Mechanisms
 , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Vitamin B12 Micronutrient: As Essential as Complex
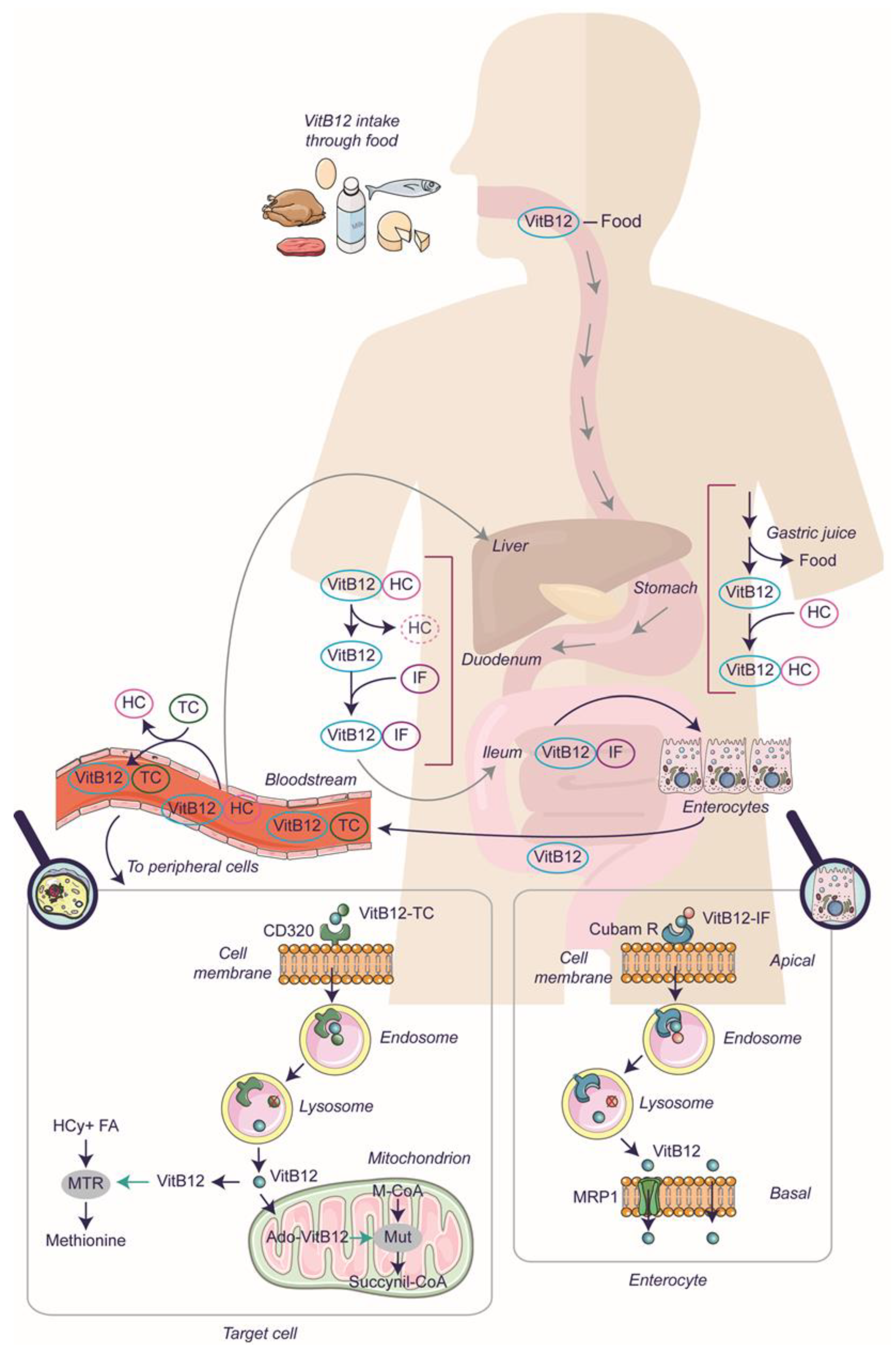
2.1. The Mechanism of Vitamin B12 Absorption
2.2. VitB12 Deficiency: The Cause and the Need for Supplementation
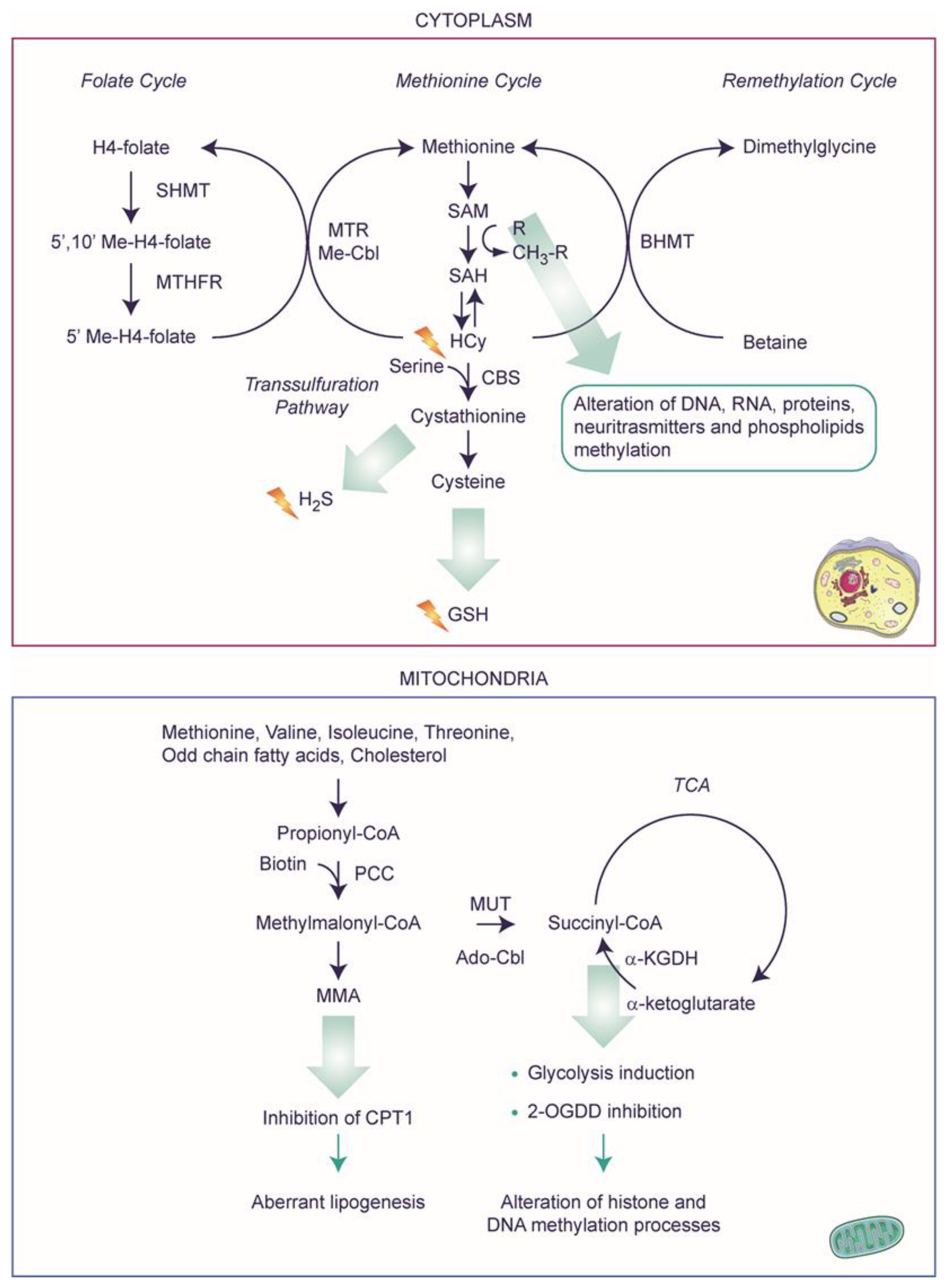
2.3. Metabolic Decompensation after VitB12 Deficiency
2.4. A Speculative Scenario of VitB12 Deficiency
3. Vitamin B12 Deficiency: What Can We Learn from Experimental Models?
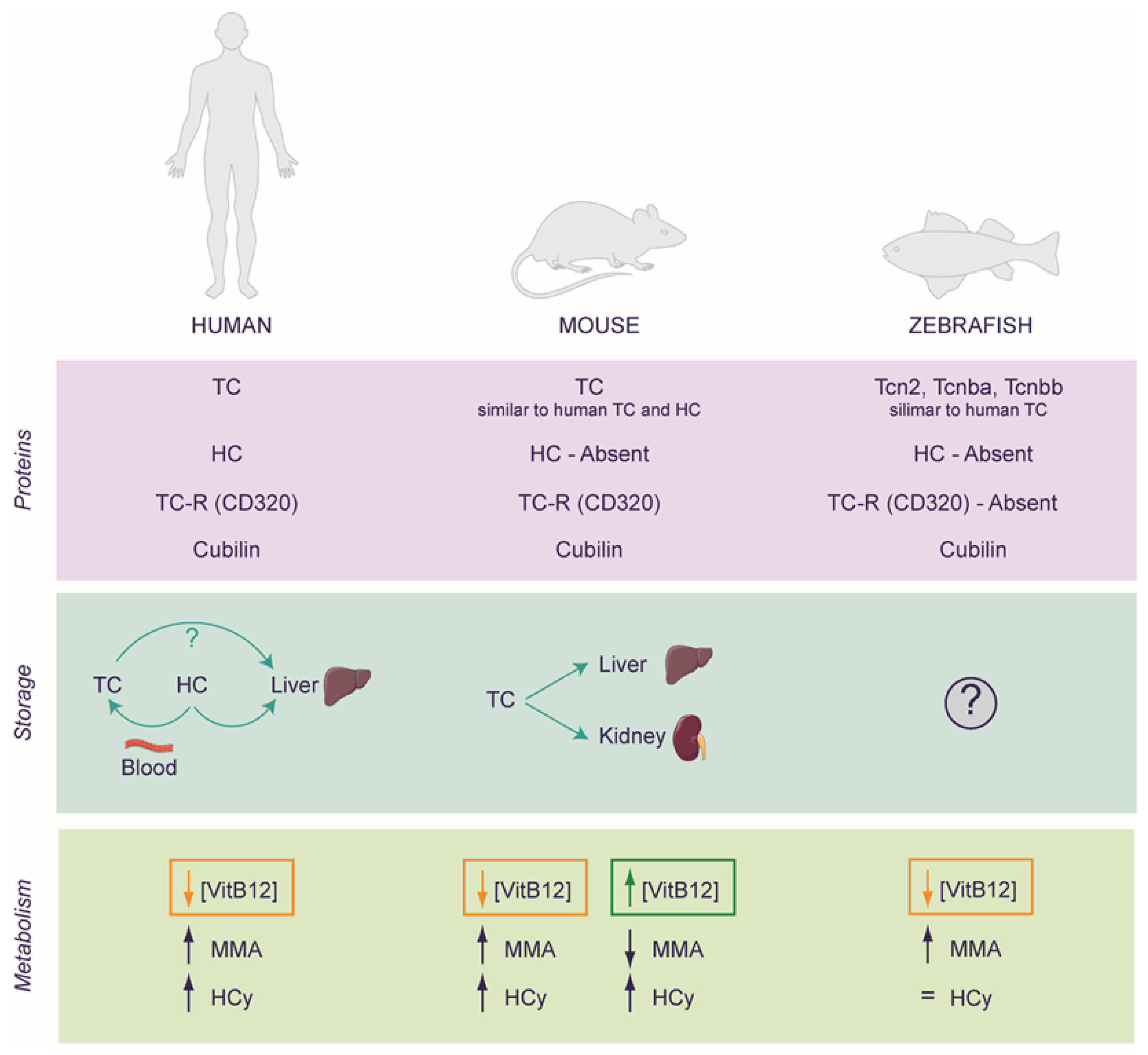
3.1. VitB12 Deficiency and Supplementation in Experimental Models: Far from Simple
3.2. Rodents as a Model for Studying VitB12 Deficiency
3.3. Zebrafish as a Model for Studying VitB12 Deficiency
4. Vitamin B12 Deficiency and Nervous System: Still Lacking Crucial Knowledge
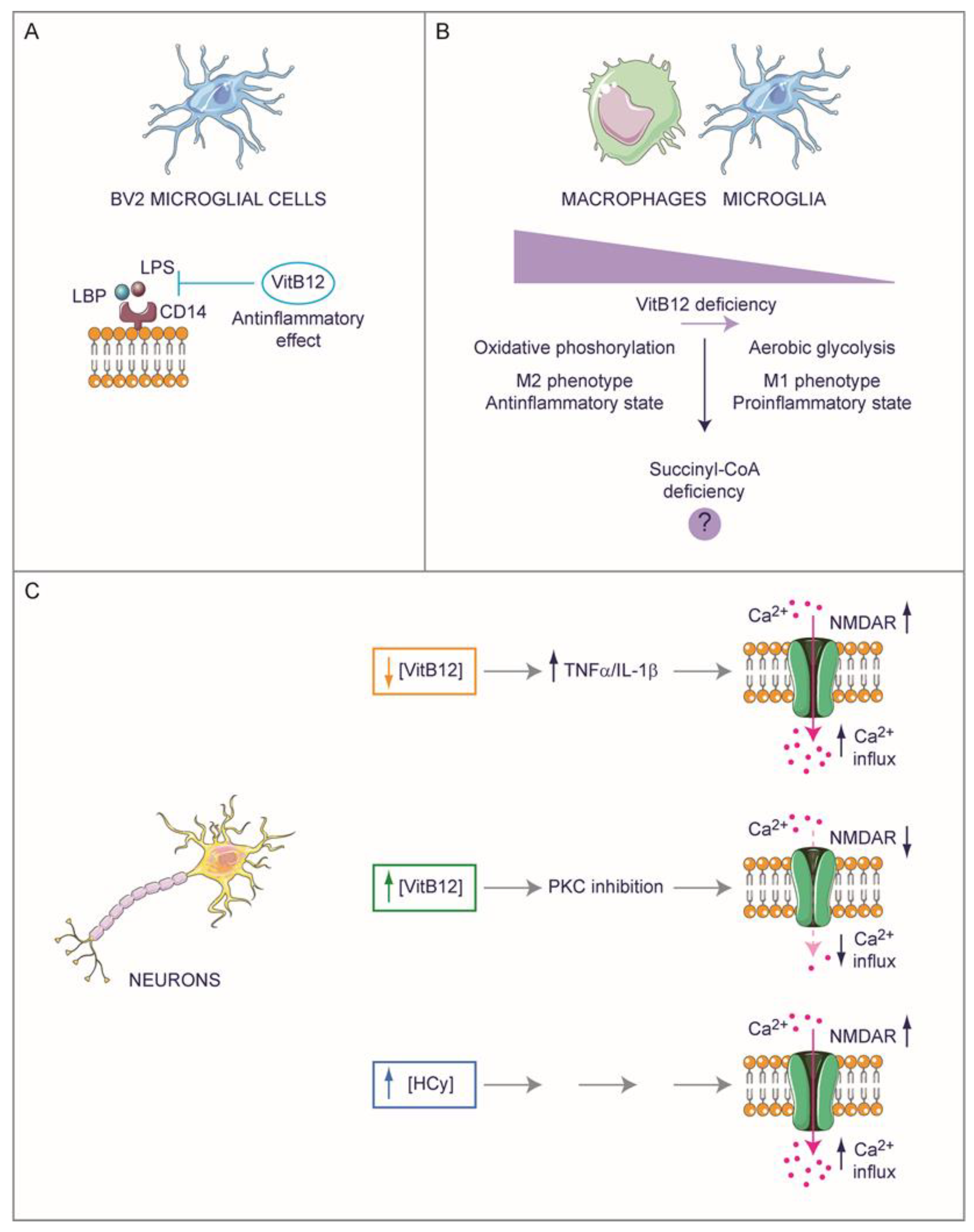
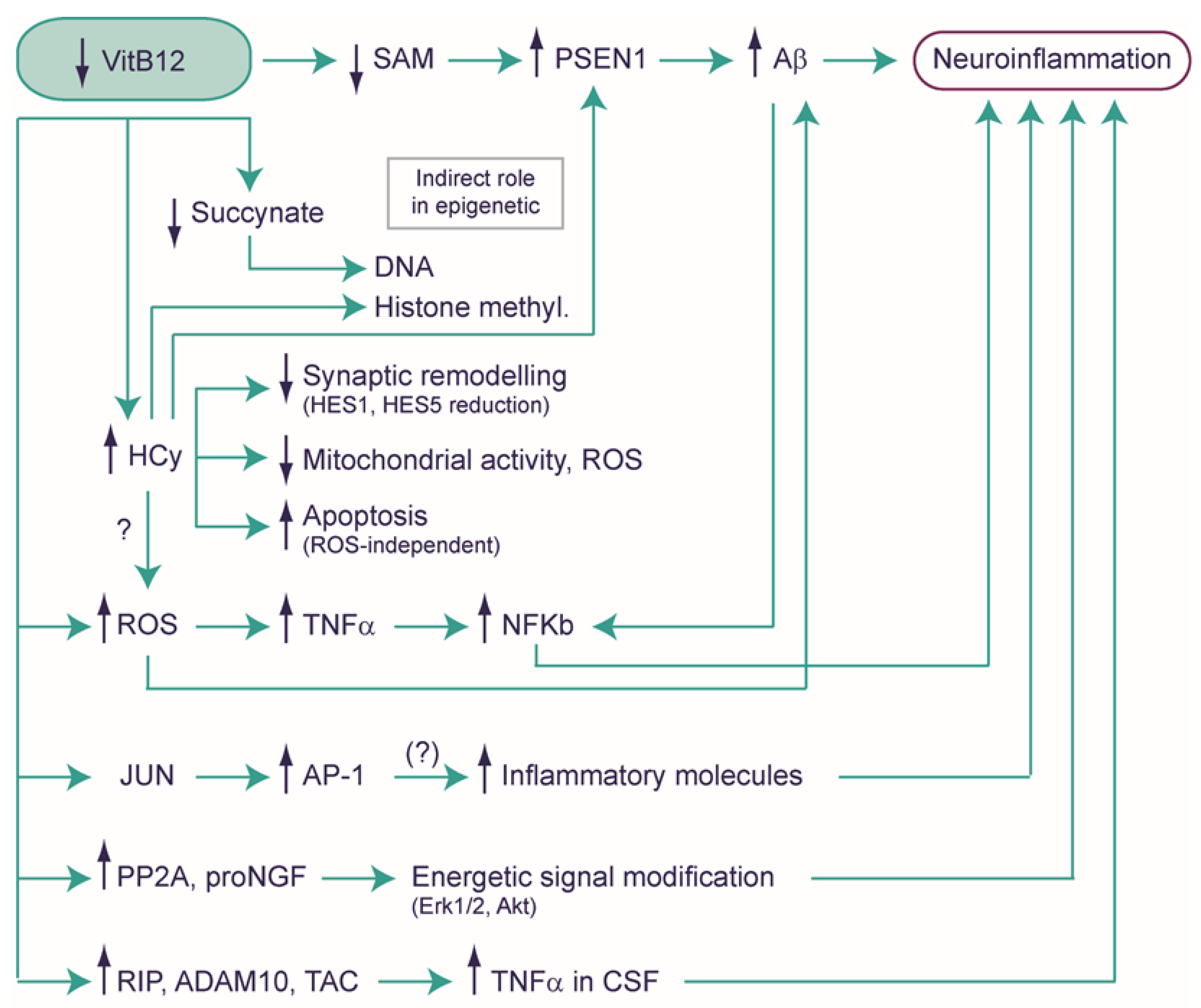
4.1. Exploring the Potential Role of Vitamin B12 in Neuroinflammation
4.2. Hyperhomocysteinemia and Neuropathology
4.3. A Possible Role of VitB12 in the Modulation of Gene Expression
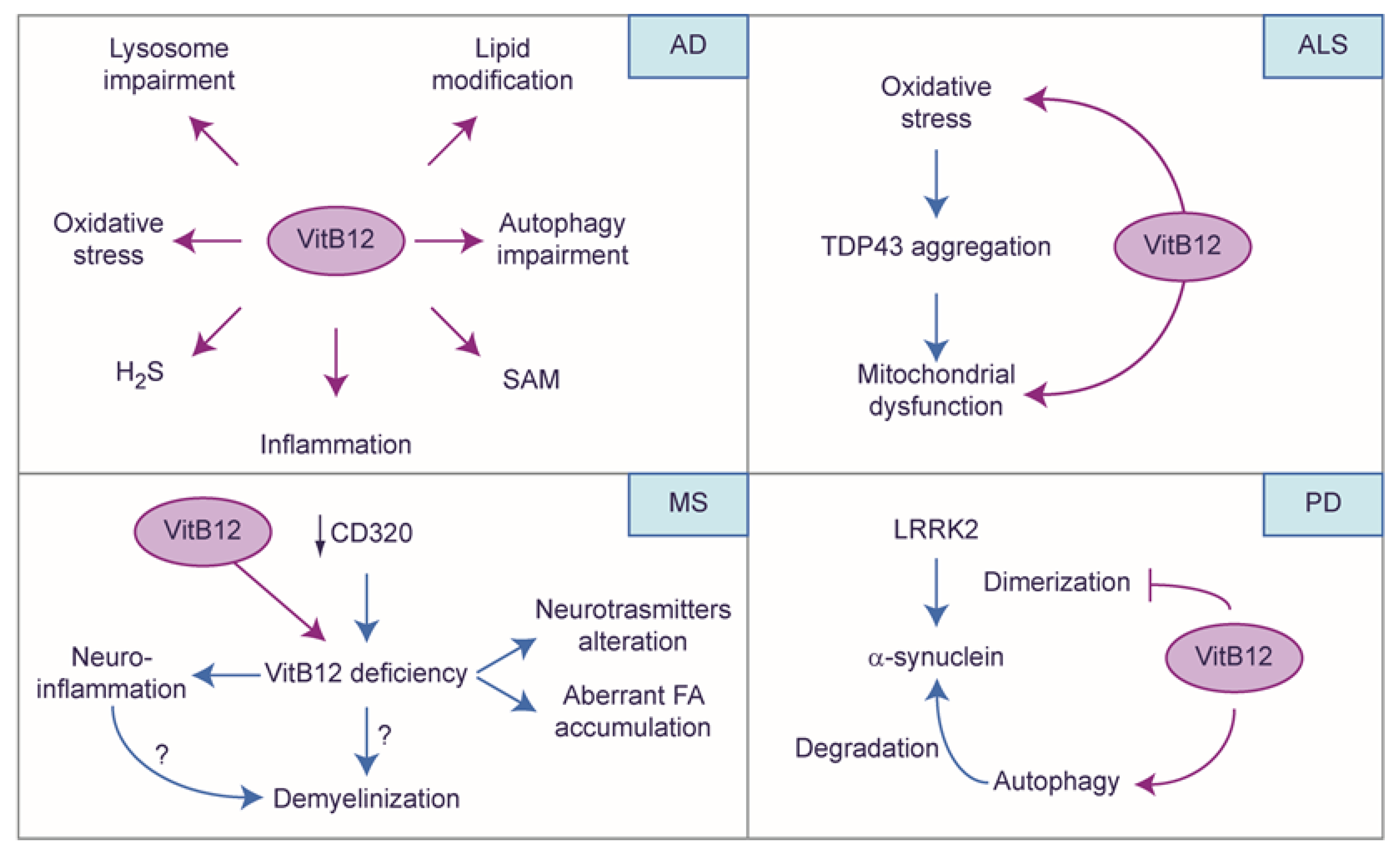
4.4. The Protective Role of Vitamin B12: A Story Told by Neuronal Diseases
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watanabe, F. Vitamin B12 Sources and Bioavailability. Exp. Biol. Med. 2007, 232, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Molloy, A.M. The Discovery of Vitamin B12. Ann. Nutr. Metab. 2012, 61, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.H. Vitamin B-12. Adv. Nutr. 2012, 3, 54–55. [Google Scholar] [CrossRef] [PubMed]
- Kräutler, B. Biochemistry of B12-Cofactors in Human Metabolism. In Sub-Cellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2012; Volume 56, pp. 323–346. [Google Scholar]
- Andrès, E.; Federici, L.; Affenberger, S.; Vidal-Alaball, J.; Loukili, N.H.; Zimmer, J.; Kaltenbach, G. B12 Deficiency: A Look beyond Pernicious Anemia. J. Fam. Pract. 2007, 56, 537–542. [Google Scholar] [PubMed]
- Ata, F.; Bint I Bilal, A.; Javed, S.; Shabir Chaudhry, H.; Sharma, R.; Fatima Malik, R.; Choudry, H.; Bhaskaran Kartha, A. Optic Neuropathy as a Presenting Feature of Vitamin B-12 Deficiency: A Systematic Review of Literature and a Case Report. Ann. Med. Surg. 2020, 60, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Sangle, P.; Sandhu, O.; Aftab, Z.; Anthony, A.T.; Khan, S. Vitamin B12 Supplementation: Preventing Onset and Improving Prognosis of Depression. Cureus 2020, 12, e11169. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P. Vitamin B 12 Deficiency. N. Engl. J. Med. 2013, 368, 149–160. [Google Scholar] [CrossRef]
- Kayali, S.; Gökcebay, D.G. Influence of Vitamin B12 Deficiency on Autonomic Nervous System Activity in Children. Iran. J. Pediatr. 2019, 29, e92634. [Google Scholar] [CrossRef]
- Scalabrino, G.; Mutti, E.; Veber, D.; Aloe, L.; Corsi, M.M.; Galbiati, S.; Tredici, G. Increased Spinal Cord NGF Levels in Rats with Cobalamin (Vitamin B12) Deficiency. Neurosci. Lett. 2006, 396, 153–158. [Google Scholar] [CrossRef]
- Henríquez, P.; Doreste, J.; Deulofeu, R.; Fiuza, M.D.; Serra-Majem, L. Nutritional Determinants of Plasma Total Homocysteine Distribution in the Canary Islands. Eur. J. Clin. Nutr. 2007, 61, 111–118. [Google Scholar] [CrossRef]
- Watkins, D.; Rosenblatt, D.S. Inborn Errors of Cobalamin Absorption and Metabolism. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Trefz, F.K.; Scheible, D.; Frauendienst-Egger, G.; Huemer, M.; Suomala, T.; Fowler, B.; Haas, D.; Baumgartner, M.R. Successful Intrauterine Treatment of a Patient with Cobalamin C Defect. Mol. Genet. Metab. Rep. 2016, 6, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Chalouhi, C.; Faesch, S.; Anthoine-Milhomme, M.-C.; Fulla, Y.; Dulac, O.; Chéron, G. Neurological Consequences of Vitamin B12 Deficiency and Its Treatment. Pediatr. Emerg. Care 2008, 24, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Gramer, G.; Hoffmann, G.F. Vitamin B(12) Deficiency in Newborns and Their Mothers-Novel Approaches to Early Detection, Treatment and Prevention of a Global Health Issue. Curr. Med. Sci. 2020, 40, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L. Chemistry and Enzymology of Vitamin B 12. Chem. Rev. 2005, 105, 2075–2150. [Google Scholar] [CrossRef] [PubMed]
- Osman, D.; Cooke, A.; Young, T.R.; Deery, E.; Robinson, N.J.; Warren, M.J. The Requirement for Cobalt in Vitamin B(12): A Paradigm for Protein Metalation. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118896. [Google Scholar] [CrossRef] [PubMed]
- Hygum, K.; Lildballe, D.L.; Greibe, E.H.; Morkbak, A.L.; Poulsen, S.S.; Sorensen, B.S.; Petersen, T.E.; Nexo, E. Mouse Transcobalamin Has Features Resembling Both Human Transcobalamin and Haptocorrin. PLoS ONE 2011, 6, e20638. [Google Scholar] [CrossRef]
- Fyfe, J.C.; Madsen, M.; Højrup, P.; Christensen, E.I.; Tanner, S.M.; de la Chapelle, A.; He, Q.; Moestrup, S.K. The Functional Cobalamin (Vitamin B12)–Intrinsic Factor Receptor Is a Novel Complex of Cubilin and Amnionless. Blood 2004, 103, 1573–1579. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Rasmussen, M.R.; Andersen, C.B.F.; Nexø, E.; Moestrup, S.K. Vitamin B 12 Transport from Food to the Body’s Cells—A Sophisticated, Multistep Pathway. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 345–354. [Google Scholar] [CrossRef]
- Kumar, S.; Mahto, M. Cerebral White Matter Demyelination in Vitamin B12 Deficiency: A Case Report. Indian J. Clin. Med. 2023, 13, 39–42. [Google Scholar] [CrossRef]
- Coelho, D.; Kim, J.C.; Miousse, I.R.; Fung, S.; Du Moulin, M.; Buers, I.; Suormala, T.; Burda, P.; Frapolli, M.; Stucki, M.; et al. Mutations in ABCD4 Cause a New Inborn Error of Vitamin B12 Metabolism. Nat. Genet. 2012, 44, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Laganà, A.S. A Review of Vitamin B12. Mol. Nutr. Vitam. 2020, 105–129. [Google Scholar] [CrossRef]
- Rashid, S.; Meier, V.; Patrick, H. Review of Vitamin B12 Deficiency in Pregnancy: A Diagnosis Not to Miss as Veganism and Vegetarianism Become More Prevalent. Eur. J. Haematol. 2021, 106, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, V.; Lenaers, G.; Urbanski, G. Diagnostic and Therapeutic Perspectives Associated to Cobalamin-Dependent Metabolism and Transcobalamins’ Synthesis in Solid Cancers. Nutrients 2022, 14, 2058. [Google Scholar] [CrossRef] [PubMed]
- Boachie, J.; Adaikalakoteswari, A.; Goljan, I.; Samavat, J.; Cagampang, F.R.; Saravanan, P. Intracellular and Tissue Levels of Vitamin B12 in Hepatocytes Are Modulated by CD320 Receptor and TCN2 Transporter. Int. J. Mol. Sci. 2021, 22, 3089. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Allen, L.H.; Bjørke-Monsen, A.-L.; Brito, A.; Guéant, J.-L.; Miller, J.W.; Molloy, A.M.; Nexo, E.; Stabler, S.; Toh, B.-H.; et al. Vitamin B(12) Deficiency. Nat. Rev. Dis. Prim. 2017, 3, 17040. [Google Scholar] [CrossRef]
- Ankar, A.; Kumar, A. Vitamin B12 Deficiency; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Degnan, P.H.; Taga, M.E.; Goodman, A.L. Vitamin B12 as a Modulator of Gut Microbial Ecology. Cell Metab. 2014, 20, 769–778. [Google Scholar] [CrossRef]
- Dholakia, K.R.; Dharmarajan, T.S.; Yadav, D.; Oiseth, S.; Norkus, E.P.; Pitchumoni, C.S. Vitamin B12 deficiency and gastric histopathology in older patients. World J. Gastroenterol. 2005, 11, 7078–7083. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Ahuja, R.; Mutti, E.; Veber, D.; Seetharam, S.; Scalabrino, G.; Seetharam, B. Cobalamin-Mediated Regulation of Transcobalamin Receptor Levels in Rat Organs. Arch. Biochem. Biophys. 2007, 463, 128–132. [Google Scholar] [CrossRef]
- Ge, Y.; Zadeh, M.; Mohamadzadeh, M. Vitamin B12 Regulates the Transcriptional, Metabolic, and Epigenetic Programing in Human Ileal Epithelial Cells. Nutrients 2022, 14, 2825. [Google Scholar] [CrossRef]
- Ge, Y.; Zadeh, M.; Mohamadzadeh, M. Vitamin B12 Coordinates Ileal Epithelial Cell and Microbiota Functions to Resist Salmonella Infection in Mice. J. Exp. Med. 2022, 219, e20220057. [Google Scholar] [CrossRef] [PubMed]
- Lurz, E.; Horne, R.G.; Määttänen, P.; Wu, R.Y.; Botts, S.R.; Li, B.; Rossi, L.; Johnson-Henry, K.C.; Pierro, A.; Surette, M.G.; et al. Vitamin B12 Deficiency Alters the Gut Microbiota in a Murine Model of Colitis. Front. Nutr. 2020, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Boachie, J.; Adaikalakoteswari, A.; Samavat, J.; Saravanan, P. Low Vitamin B12 and Lipid Metabolism: Evidence from Pre-Clinical and Clinical Studies. Nutrients 2020, 12, 1925. [Google Scholar] [CrossRef] [PubMed]
- Rush, E.C.; Katre, P.; Yajnik, C.S. Vitamin B12: One Carbon Metabolism, Fetal Growth and Programming for Chronic Disease. Eur. J. Clin. Nutr. 2014, 68, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Chern, T.; Achilleos, A.; Tong, X.; Hsu, C.-W.; Wong, L.; Poché, R.A. Mouse Models to Study the Pathophysiology of Combined Methylmalonic Acidemia and Homocystinuria, CblC Type. Dev. Biol. 2020, 468, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, J.L.K.; Saenz, M.S.; Thomas, J.A.; Gallagher, R.C.; Lovell, M.A.; Fenton, L.Z.; Shanske, S.; Myers, S.M.; Wanders, R.J.A.; Ruiter, J.; et al. Succinyl-CoA Ligase Deficiency: A Mitochondrial Hepatoencephalomyopathy. Pediatr. Res. 2010, 68, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Chien, D.; Dean, D.; Saha, A.K.; Flatt, J.P.; Ruderman, N.B. Malonyl-CoA Content and Fatty Acid Oxidation in Rat Muscle and Liver in Vivo. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E259–E265. [Google Scholar] [CrossRef]
- Froese, D.S.; Gravel, R.A. Genetic Disorders of Vitamin B₁₂ Metabolism: Eight Complementation Groups—Eight Genes. Expert Rev. Mol. Med. 2010, 12, e37. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic Regulation of Microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Kramer, P.A.; Ravi, S.; Chacko, B.; Johnson, M.S.; Darley-Usmar, V.M. A Review of the Mitochondrial and Glycolytic Metabolism in Human Platelets and Leukocytes: Implications for Their Use as Bioenergetic Biomarkers. Redox Biol. 2014, 2, 206–210. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-Alpha Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in Plasma Homocysteine Associated with Parallel Increases in Plasma S-Adenosylhomocysteine and Lymphocyte DNA Hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef] [PubMed]
- van de Lagemaat, E.E.; de Groot, L.C.P.G.M.; van den Heuvel, E.G.H.M. Vitamin B(12) in Relation to Oxidative Stress: A Systematic Review. Nutrients 2019, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef]
- Lildballe, D.L.; Mutti, E.; Birn, H.; Nexo, E. Maximal Load of the Vitamin B12 Transport System: A Study on Mice Treated for Four Weeks with High-Dose Vitamin B12 or Cobinamide. PLoS ONE 2012, 7, e46657. [Google Scholar] [CrossRef]
- Mutti, E.; Ruetz, M.; Birn, H.; Kräutler, B.; Nexo, E. 4-Ethylphenyl-Cobalamin Impairs Tissue Uptake of Vitamin B12 and Causes Vitamin B12 Deficiency in Mice. PLoS ONE 2013, 8, e75312. [Google Scholar] [CrossRef]
- Chern, T.; Achilleos, A.; Tong, X.; Hill, M.C.; Saltzman, A.B.; Reineke, L.C.; Chaudhury, A.; Dasgupta, S.K.; Redhead, Y.; Watkins, D.; et al. Mutations in Hcfc1 and Ronin Result in an Inborn Error of Cobalamin Metabolism and Ribosomopathy. Nat. Commun. 2022, 13, 134. [Google Scholar] [CrossRef]
- Buccellato, F.R.; Miloso, M.; Braga, M.; Nicolini, G.; Morabito, A.; Pravettoni, G.; Tredici, G.; Scalabrino, G. Myelinolytic Lesions in Spinal Cord of Cobalamin-deficient Rats Are TNF-α-mediated. FASEB J. 1999, 13, 297–304. [Google Scholar] [CrossRef]
- Dinn, J.; Mccann, S.; Wilson, P.; Reed, B.; Weir, D.; Scott, J. Animal Model For Subacute Combined Degeneration. Lancet 1978, 312, 1154. [Google Scholar] [CrossRef]
- Paz, D.; Pinales, B.E.; Castellanos, B.S.; Perez, I.; Gil, C.B.; Madrigal, L.J.; Reyes-Nava, N.G.; Castro, V.L.; Sloan, J.L.; Quintana, A.M. Abnormal Chondrocyte Development in a Zebrafish Model of CblC Syndrome Restored by an MMACHC Cobalamin Binding Mutant. Differentiation 2023, 131, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.L.; Achilly, N.P.; Arnold, M.L.; Catlett, J.L.; Blake, T.; Bishop, K.; Jones, M.; Harper, U.; English, M.A.; Anderson, S.; et al. The Vitamin B12 Processing Enzyme, Mmachc, Is Essential for Zebrafish Survival, Growth and Retinal Morphology. Hum. Mol. Genet. 2020, 29, 2109–2123. [Google Scholar] [CrossRef] [PubMed]
- Arigony, A.L.V.; de Oliveira, I.M.; Machado, M.; Bordin, D.L.; Bergter, L.; Prá, D.; Henriques, J.A.P. The Influence of Micronutrients in Cell Culture: A Reflection on Viability and Genomic Stability. Biomed. Res. Int. 2013, 2013, 597282. [Google Scholar] [CrossRef]
- Ermens, A.A.M.; Vlasveld, L.T.; Lindemans, J. Significance of Elevated Cobalamin (Vitamin B12) Levels in Blood. Clin. Biochem. 2003, 36, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R. Biomarkers of Cobalamin (Vitamin B-12) Status in the Epidemiologic Setting: A Critical Overview of Context, Applications, and Performance Characteristics of Cobalamin, Methylmalonic Acid, and Holotranscobalamin II. Am. J. Clin. Nutr. 2011, 94, 348S–358S. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Tacconi, S.; Sbarigia, C.; Passeri, D.; Rossi, M.; Tata, A.M.; Dini, L. Current Nanocarrier Strategies Improve Vitamin B12 Pharmacokinetics, Ameliorate Patients’ Lives, and Reduce Costs. Nanomaterials 2021, 11, 743. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. In Neuronal Cell Culture: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2013; pp. 9–21. [Google Scholar]
- Dastidar, R.; Sikder, K. Diagnostic Reliability of Serum Active B12 (Holo-Transcobalamin) in True Evaluation of Vitamin B12 Deficiency: Relevance in Current Perspective. BMC Res. Notes 2022, 15, 329. [Google Scholar] [CrossRef]
- Beedholm-Ebsen, R.; van de Wetering, K.; Hardlei, T.; Nexø, E.; Borst, P.; Moestrup, S.K. Identification of Multidrug Resistance Protein 1 (MRP1/ABCC1) as a Molecular Gate for Cellular Export of Cobalamin. Blood 2010, 115, 1632–1639. [Google Scholar] [CrossRef]
- Tamura, J.; Kubota, K.; Murakami, H.; Sawamura, M.; Matsushima, T.; Tamura, T.; Saitoh, T.; Kurabayshi, H.; Naruse, T. Immunomodulation by Vitamin B12: Augmentation of CD8+ T Lymphocytes and Natural Killer (NK) Cell Activity in Vitamin B12-Deficient Patients by Methyl-B12 Treatment. Clin. Exp. Immunol. 1999, 116, 28–32. [Google Scholar] [CrossRef]
- Jonnalagadda, D.; Kihara, Y.; Groves, A.; Ray, M.; Saha, A. FTY720 Requires Vitamin B 12 -TCN2-CD320 Signaling in Astrocytes to Reduce Disease in an Animal Model of Multiple Sclerosis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zhao, H.; Ruberu, K.; Li, H.; Garner, B. Analysis of Subcellular [57Co] Cobalamin Distribution in SH-SY5Y Neurons and Brain Tissue. J. Neurosci. Methods 2013, 217, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, H.; Ruberu, K.; Garner, B. Impaired Lysosomal Cobalamin Transport in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 43, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Benoit, C.R.; Stanton, A.E.; Tartanian, A.C.; Motzer, A.R.; McGaughey, D.M.; Bond, S.R.; Brody, L.C. Functional and phylogenetic characterization of noncanonical vitamin B12-binding proteins in zebrafish suggests involvement in cobalamin transport. J. Biol. Chem. 2018, 293, 17606–17621. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Ellis, J.P.; Tirone, J.C.; Pawelek, P.D.; Doré, C.; Atkinson, J.L.; Watkins, D.; Morel, C.F.; Fujiwara, T.M.; Moras, E.; Hosack, A.R.; et al. Identification of the Gene Responsible for Methylmalonic Aciduria and Homocystinuria, CblC Type. Nat. Genet. 2006, 38, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Carrasco, N.; Chandler, R.J.; Venditti, C.P. Combined Methylmalonic Acidemia and Homocystinuria, CblC Type. I. Clinical Presentations, Diagnosis and Management. J. Inherit. Metab. Dis. 2012, 35, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Deodato, F.; Dionisi-Vici, C. Cobalamin C Defect: Natural History, Pathophysiology, and Treatment. J. Inherit. Metab. Dis. 2011, 34, 127–135. [Google Scholar] [CrossRef]
- Weisfeld-Adams, J.D.; McCourt, E.A.; Diaz, G.A.; Oliver, S.C. Ocular Disease in the Cobalamin C Defect: A Review of the Literature and a Suggested Framework for Clinical Surveillance. Mol. Genet. Metab. 2015, 114, 537–546. [Google Scholar] [CrossRef]
- Plesa, M.; Kim, J.; Paquette, S.G.; Gagnon, H.; Ng-Thow-Hing, C.; Gibbs, B.F.; Hancock, M.A.; Rosenblatt, D.S.; Coulton, J.W. Interaction between MMACHC and MMADHC, Two Human Proteins Participating in Intracellular Vitamin B₁₂ Metabolism. Mol. Genet. Metab. 2011, 102, 139–148. [Google Scholar] [CrossRef]
- Froese, D.S.; Kopec, J.; Fitzpatrick, F.; Schuller, M.; McCorvie, T.J.; Chalk, R.; Plessl, T.; Fettelschoss, V.; Fowler, B.; Baumgartner, M.R.; et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J. Biol. Chem. 2015, 290, 29167–29177. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, L.; Chen, Q.; Wang, Y.; Peng, Y.; Tang, H. Case Report: A Case of Late-Onset Combined Methylmalonic Acidemia and Hyperhomocysteinemia Induced by a Vegetarian Diet. Front. Pediatr. 2022, 10, 896177. [Google Scholar] [CrossRef]
- Wang, X.; Sun, W.; Yang, Y.; Jia, J.; Li, C. A Clinical and Gene Analysis of Late-Onset Combined Methylmalonic Aciduria and Homocystinuria, CblC Type, in China. J. Neurol. Sci. 2012, 318, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.C.-H.; Morel, C.F.; Scharer, G.; Yang, M.; Lerner-Ellis, J.P.; Rosenblatt, D.S.; Thomas, J.A. Late-Onset Combined Homocystinuria and Methylmalonic Aciduria (CblC) and Neuropsychiatric Disturbance. Am. J. Med. Genet. A 2007, 143A, 2430–2434. [Google Scholar] [CrossRef] [PubMed]
- Brox-Torrecilla, N.; Arhip, L.; Miguélez-González, M.; Castellano-Gasch, S.; Contreras-Chicote, A.; Rodríguez-Ferrero, M.L.; Motilla de la Cámara, M.L.; Serrano-Moreno, C.; Cuerda Compes, C. Late-Onset Methylmalonic Acidemia and Homocysteinemia. Nutr. Hosp. 2021, 38, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-Y.; An, H.; Liu, J.; Li, J.-Y.; Han, Y.; Zhou, A.-H.; Wang, F.; Jia, J.-P. Manic-Depressive Psychosis as the Initial Symptom in Adult Siblings with Late-Onset Combined Methylmalonic Aciduria and Homocystinemia, Cobalamin C Type. Chin. Med. J. 2017, 130, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Huemer, M.; Scholl-Bürgi, S.; Hadaya, K.; Kern, I.; Beer, R.; Seppi, K.; Fowler, B.; Baumgartner, M.R.; Karall, D. Three New Cases of Late-Onset CblC Defect and Review of the Literature Illustrating When to Consider Inborn Errors of Metabolism beyond Infancy. Orphanet J. Rare Dis. 2014, 9, 161. [Google Scholar] [CrossRef]
- Pepper, M.R.; Black, M.M. B12 in Fetal Development. Semin. Cell Dev. Biol. 2011, 22, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Rodríguez, J.; Díaz-López, A.; Canals-Sans, J.; Arija, V. Maternal Vitamin B12 Status during Pregnancy and Early Infant Neurodevelopment: The ECLIPSES Study. Nutrients 2023, 15, 1529. [Google Scholar] [CrossRef]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O.; Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the Nervous System: Current Knowledge of the Biochemical Modes of Action and Synergies of Thiamine, Pyridoxine, and Cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef]
- Rakić, M.; Lunić, T.; Bekić, M.; Tomić, S.; Mitić, K.; Graovac, S.; Božić, B.; Božić Nedeljković, B. Vitamin B Complex Suppresses Neuroinflammation in Activated Microglia: In Vitro and in Silico Approach Combined with Dynamical Modeling. Int. Immunopharmacol. 2023, 121, 110525. [Google Scholar] [CrossRef]
- Fuso, A.; Cavallaro, R.A.; Orrù, L.; Buttarelli, F.R.; Scarpa, S. Gene Silencing by S-Adenosylmethionine in Muscle Differentiation. FEBS Lett. 2001, 508, 337–340. [Google Scholar] [CrossRef]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-Adenosylmethionine/Homocysteine Cycle Alterations Modify DNA Methylation Status with Consequent Deregulation of PS1 and BACE and Beta-Amyloid Production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 Gene Silencing by S-Adenosylmethionine: A Treatment for Alzheimer Disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ali, R.; Verma, S. Aβ-Oligomers: A Potential Therapeutic Target for Alzheimer’s Disease. Int. J. Biol. Macromol. 2023, 239, 124231. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Cavallaro, R.A.; Nicolia, V.; Scarpa, S. PSEN1 Promoter Demethylation in Hyperhomocysteinemic TgCRND8 Mice Is the Culprit, Not the Consequence. Curr. Alzheimer Res. 2012, 9, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-E.; Wei, W.; Liu, Y.-H.; Peng, J.-H.; Tian, Q.; Liu, G.-P.; Zhang, Y.; Wang, J.-Z. Hyperhomocysteinemia Increases Beta-Amyloid by Enhancing Expression of Gamma-Secretase and Phosphorylation of Amyloid Precursor Protein in Rat Brain. Am. J. Pathol. 2009, 174, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Prajjwal, P.; Asharaf, S.; Makhanasa, D.; Yamparala, A.; Tariq, H.; Aleti, S.; Gadam, S.; Vora, N. Association of Alzheimer’s Dementia with Oral Bacteria, Vitamin B12, Folate, Homocysteine Levels, and Insulin Resistance along with Its Pathophysiology, Genetics, Imaging, and Biomarkers. Dis. Mon. 2023, 69, 101546. [Google Scholar] [CrossRef]
- Alam, P.; Siddiqi, M.K.; Chaturvedi, S.K.; Zaman, M.; Khan, R.H. Vitamin B12 Offers Neuronal Cell Protection by Inhibiting Aβ-42 Amyloid Fibrillation. Int. J. Biol. Macromol. 2017, 99, 477–482. [Google Scholar] [CrossRef]
- Andrade, S.; Loureiro, J.A.; Pereira, M.C. The Role of Amyloid β-Biomembrane Interactions in the Pathogenesis of Alzheimer’s Disease: Insights from Liposomes as Membrane Models. Chemphyschem 2021, 22, 1547–1565. [Google Scholar] [CrossRef]
- Peracchi, M.; Bamonti Catena, F.; Pomati, M.; De Franceschi, M.; Scalabrino, G. Human Cobalamin Deficiency: Alterations in Serum Tumour Necrosis Factor-Alpha and Epidermal Growth Factor. Eur. J. Haematol. 2001, 67, 123–127. [Google Scholar] [CrossRef]
- Veber, D.; Mutti, E.; Tacchini, L.; Gammella, E.; Tredici, G.; Scalabrino, G. Indirect Down-Regulation of Nuclear NF-KappaB Levels by Cobalamin in the Spinal Cord and Liver of the Rat. J. Neurosci. Res. 2008, 86, 1380–1387. [Google Scholar] [CrossRef]
- Scalabrino, G.; Buccellato, F.R.; Veber, D.; Mutti, E. New Basis of the Neurotrophic Action of Vitamin B12. Clin. Chem. Lab. Med. 2003, 41, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- Scalabrino, G.; Galimberti, D.; Mutti, E.; Scalabrini, D.; Veber, D.; De Riz, M.; Bamonti, F.; Capello, E.; Mancardi, G.L.; Scarpini, E.; et al. Loss of Epidermal Growth Factor Regulation by Cobalamin in Multiple Sclerosis. Brain Res. 2010, 1333, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mutti, E.; Magnaghi, V.; Veber, D.; Faroni, A.; Pece, S.; Di Fiore, P.P.; Scalabrino, G. Cobalamin Deficiency-Induced Changes of Epidermal Growth Factor (EGF)-Receptor Expression and EGF Levels in Rat Spinal Cord. Brain Res. 2011, 1376, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ciappio, E.D.; Crott, J.W.; Brooks, R.S.; Nesvet, J.; Smith, D.E.; Choi, S.-W.; Mason, J.B. Combined Inadequacies of Multiple B Vitamins Amplify Colonic Wnt Signaling and Promote Intestinal Tumorigenesis in BAT-LacZxApc1638N Mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3136–3145. [Google Scholar] [CrossRef]
- Wei, C.-J.; Li, Y.-L.; Zhu, Z.-L.; Jia, D.-M.; Fan, M.-L.; Li, T.; Wang, X.-J.; Li, Z.-G.; Ma, H.-S. Inhibition of Activator Protein 1 Attenuates Neuroinflammation and Brain Injury after Experimental Intracerebral Hemorrhage. CNS Neurosci. Ther. 2019, 25, 1182–1188. [Google Scholar] [CrossRef]
- Battaglia-Hsu, S.-F.; Akchiche, N.; Noel, N.; Alberto, J.M.; Jeannesson, E.; Orozco-Barrios, C.E.; Martinez-Fong, D.; Daval, J.L.; Guéant, J.L. Vitamin B12 Deficiency Reduces Proliferation and Promotes Differentiation of Neuroblastoma Cells and Up-Regulates PP2A, ProNGF, and TACE. Proc. Natl. Acad. Sci. USA 2009, 106, 21930–21935. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Hung, K.-L.; Wang, C.-C.; Huang, C.-Y.; Wang, S.-J. Cyanocobalamin, Vitamin B12, Depresses Glutamate Release through Inhibition of Voltage-Dependent Ca2+ Influx in Rat Cerebrocortical Nerve Terminals (Synaptosomes). Eur. J. Pharmacol. 2009, 602, 230–237. [Google Scholar] [CrossRef]
- Liu, H.; Wen, L.-M.; Qiao, H.; An, S.-C. Modulation of hippocampal glutamate and NMDA/AMPA receptor by homocysteine in chronic unpredictable mild stress-induced rat depression. Sheng Li Xue Bao 2013, 65, 61–71. [Google Scholar]
- Chen, B.-S.; Roche, K.W. Regulation of NMDA Receptors by Phosphorylation. Neuropharmacology 2007, 53, 362–368. [Google Scholar] [CrossRef]
- Roth, W.; Mohamadzadeh, M. Vitamin B12 and Gut-Brain Homeostasis in the Pathophysiology of Ischemic Stroke. EBioMedicine 2021, 73, 103676. [Google Scholar] [CrossRef] [PubMed]
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxid. Redox Signal. 2011, 14, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Guest, J.; Bilgin, A.; Hokin, B.; Mori, T.A.; Croft, K.D.; Grant, R. Novel Relationships between B12, Folate and Markers of Inflammation, Oxidative Stress and NAD(H) Levels, Systemically and in the CNS of a Healthy Human Cohort. Nutr. Neurosci. 2015, 18, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-W.; Ma, Y.-M.; Jing, L.; Wang, Y.-L.; Zhang, J.-Z. Synaptic Remodeling and Reduced Expression of the Transcription Factors, HES1 and HES5, in the Cortex Neurons of Cognitively Impaired Hyperhomocysteinemic Mice. Pathol. Res. Pract. 2020, 216, 152953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Huang, D.; Hou, J.; Li, J.; Zhang, Y.; Tian, M.; Li, Z.; Tie, T.; Cheng, Y.; Su, X.; et al. High-Concentration Homocysteine Inhibits Mitochondrial Respiration Function and Production of Reactive Oxygen Species in Neuron Cells. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2020, 29, 105109. [Google Scholar] [CrossRef] [PubMed]
- Wyse, A.T.S.; Sanches, E.F.; Dos Santos, T.M.; Siebert, C.; Kolling, J.; Netto, C.A. Chronic Mild Hyperhomocysteinemia Induces Anxiety-like Symptoms, Aversive Memory Deficits and Hippocampus Atrophy in Adult Rats: New Insights into Physiopathological Mechanisms. Brain Res. 2020, 1728, 146592. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Roh, H.; Kwon, Y. Causes of Hyperhomocysteinemia and Its Pathological Significance. Arch. Pharm. Res. 2018, 41, 372–383. [Google Scholar] [CrossRef]
- Molognoni, F.; de Melo, F.H.M.; da Silva, C.T.; Jasiulionis, M.G. Ras and Rac1, Frequently Mutated in Melanomas, Are Activated by Superoxide Anion, Modulate Dnmt1 Level and Are Causally Related to Melanocyte Malignant Transformation. PLoS ONE 2013, 8, e81937. [Google Scholar] [CrossRef]
- de Queiroz, K.B.; Cavalcante-Silva, V.; Lopes, F.L.; Rocha, G.A.; D’Almeida, V.; Coimbra, R.S. Vitamin B(12) Is Neuroprotective in Experimental Pneumococcal Meningitis through Modulation of Hippocampal DNA Methylation. J. Neuroinflammation 2020, 17, 96. [Google Scholar] [CrossRef]
- Mathew, A.R.; Cavallucci, V.; Fidaleo, M. Altered Vitamin B12 Metabolism in the Central Nervous System Is Associated with the Modification of Ribosomal Gene Expression: New Insights from Comparative RNA Dataset Analysis. Funct. Integr. Genom. 2023, 23, 45. [Google Scholar] [CrossRef]
- Zhong, L.; Zhou, J.; Chen, X.; Lou, Y.; Liu, D.; Zou, X.; Yang, B.; Yin, Y.; Pan, Y. Quantitative Proteomics Study of the Neuroprotective Effects of B12 on Hydrogen Peroxide-Induced Apoptosis in SH-SY5Y Cells. Sci. Rep. 2016, 6, 22635. [Google Scholar] [CrossRef] [PubMed]
- Birch, C.S.; Brasch, N.E.; McCaddon, A.; Williams, J.H.H. A Novel Role for Vitamin B(12): Cobalamins Are Intracellular Antioxidants in Vitro. Free Radic. Biol. Med. 2009, 47, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Requejo-Aguilar, R.; Gonzalez-Fernandez, S.; Resch, M.; Carabias-Carrasco, M.; Ros, J.; Almeida, A.; Bolaños, J.P. Hippocampal Neurons Require a Large Pool of Glutathione to Sustain Dendrite Integrity and Cognitive Function. Redox Biol. 2018, 19, 52–61. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. A Diet Rich in Taurine, Cysteine, Folate, B(12) and Betaine May Lessen Risk for Alzheimer’s Disease by Boosting Brain Synthesis of Hydrogen Sulfide. Med. Hypotheses 2019, 132, 109356. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.W.; Grösgen, S.; Riemenschneider, M.; Tanila, H.; Grimm, H.S.; Hartmann, T. From Brain to Food: Analysis of Phosphatidylcholins, Lyso-Phosphatidylcholins and Phosphatidylcholin-Plasmalogens Derivates in Alzheimer’s Disease Human Post Mortem Brains and Mice Model via Mass Spectrometry. J. Chromatogr. A 2011, 1218, 7713–7722. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.J.; Mancera, R.L.; Howard, M.; Arrigan, D.W.M.; Newsholme, P.; Martins, R.N. Role of the Cell Membrane Interface in Modulating Production and Uptake of Alzheimer’s Beta Amyloid Protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1639–1651. [Google Scholar] [CrossRef]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma Nutrient Status of Patients with Alzheimer’s Disease: Systematic Review and Meta-Analysis. Alzheimer’s Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H.-F. Associations between Homocysteine, Folic Acid, Vitamin B12 and Alzheimer’s Disease: Insights from Meta-Analyses. J. Alzheimer’s. Dis. 2015, 46, 777–790. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Lütjohann, D.; von Bergmann, K.; Verhey, F.; Vreeling, F.; Wauters, A.; Bosmans, E.; Bosma, H.; van Boxtel, M.P.J.; Maes, M.; et al. Combination of Serum Markers Related to Several Mechanisms in Alzheimer’s Disease. Neurobiol. Aging 2003, 24, 893–902. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Ge, B.; Zhou, D.; Li, M.; Li, W.; Ma, F.; Liu, Z.; Ji, Y.; Huang, G. Effects of Folic Acid and Vitamin B12 Supplementation on Cognitive Impairment and Inflammation in Patients with Alzheimer’s Disease: A Randomized, Single-Blinded, Placebo-Controlled Trial. J. Prev. Alzheimer’s Dis. 2021, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Doshanjh, L.; Fishman, P.; Luo, Y.; Smyers, K.; Page, R.; Morrell, C.; et al. A Phase II Randomized Clinical Trial of a Nutritional Formulation for Cognition and Mood in Alzheimer’s Disease. J. Alzheimer’s. Dis. 2015, 45, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Theiss, E.L.; Griebsch, L.V.; Lauer, A.A.; Janitschke, D.; Erhardt, V.K.J.; Haas, E.C.; Kuppler, K.N.; Radermacher, J.; Walzer, O.; Portius, D.; et al. Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells. Cells 2022, 11, 2574. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Li, D.; Dong, Y.; She, Q.; Zhou, T.; Nie, X.; Pan, R.; Deng, Y. Methylcobalamin Protects Melanocytes from H2O2-Induced Oxidative Stress by Activating the Nrf2/HO-1 Pathway. Drug Des. Devel. Ther. 2021, 15, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic Targeting of the NRF2 and KEAP1 Partnership in Chronic Diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Petrillo, S.; Bertini, E.S.; Piemonte, F. Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement. Biomolecules 2020, 10, 702. [Google Scholar] [CrossRef]
- Tripathi, M.; Zhang, C.W.; Singh, B.K.; Sinha, R.A.; Moe, K.T.; DeSilva, D.A.; Yen, P.M. Hyperhomocysteinemia Causes ER Stress and Impaired Autophagy That Is Reversed by Vitamin B Supplementation. Cell Death Dis. 2016, 7, e2513. [Google Scholar] [CrossRef]
- Mehrdad, J.; Leila, E.; Emsehgol, N. The Effect of Vitamin B12 on Synaptic Plasticity of Hippocampus in Alzheimer’s Disease Model Rats. Int. J. Neurosci. 2023, 133, 654–659. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 Aggregation Induced by Oxidative Stress Causes Global Mitochondrial Imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Jeon, Y.-M.; Kwon, Y.; Lee, S.; Kim, S.; Jo, M.; Lee, S.; Kim, S.R.; Kim, K.; Kim, H.-J. Vitamin B12 Reduces TDP-43 Toxicity by Alleviating Oxidative Stress and Mitochondrial Dysfunction. Antioxidants 2021, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, A.; Li, X.; Gomez-Llorente, Y.; Leandrou, E.; Memou, A.; Clemente, N.; Yao, C.; Afsari, F.; Zhi, L.; Pan, N.; et al. Vitamin B(12) Modulates Parkinson’s Disease LRRK2 Kinase Activity through Allosteric Regulation and Confers Neuroprotection. Cell Res. 2019, 29, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Yang, X.; Chen, X.; Xiao, D.; Zhu, J.; Zhang, M.; Qin, X.; Ma, X.; Lin, Y. Treating LRRK2-Related Parkinson’s Disease by Inhibiting the MTOR Signaling Pathway to Restore Autophagy. Adv. Funct. Mater. 2021, 31, 2105152. [Google Scholar] [CrossRef]
- Arora, K.; Sequeira, J.M.; Alarcon, J.M.; Wasek, B.; Arning, E.; Bottiglieri, T.; Quadros, E. V Neuropathology of Vitamin B 12 Deficiency in the Cd320−/− Mouse. FASEB J. 2019, 33, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., III; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-Analysis: High-Dosage Vitamin E Supplementation May Increase All-Cause Mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Groves, A.; Kihara, Y.; Jonnalagadda, D.; Rivera, R.; Kennedy, G.; Mayford, M.; Chun, J. A Functionally Defined In Vivo Astrocyte Population Identified by C-Fos Activation in a Mouse Model of Multiple Sclerosis Modulated by S1P Signaling: Immediate-Early Astrocytes (IeAstrocytes). eNeuro 2018, 5, ENEURO.0239-18.2018. [Google Scholar] [CrossRef]
- Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin Deficiency: Clinical Picture and Radiological Findings. Nutrients 2013, 5, 4521–4539. [Google Scholar] [CrossRef]
- Nawaz, A.; Khattak, N.N.; Khan, M.S.; Nangyal, H.; Sabri, S.; Shakir, M. Deficiency of Vitamin B12 and Its Relation with Neurological Disorders: A Critical Review. J. Basic Appl. Zool. 2020, 81, 10. [Google Scholar] [CrossRef]
- Scalabrino, G.; Veber, D.; Mutti, E. Experimental and Clinical Evidence of the Role of Cytokines and Growth Factors in the Pathogenesis of Acquired Cobalamin-Deficient Leukoneuropathy. Brain Res. Rev. 2008, 59, 42–54. [Google Scholar] [CrossRef]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The Adult Oligodendrocyte Can Participate in Remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Tanaka, H.; Okamoto, M.; Okada, K.; Murase, T.; Yoshikawa, H. Methylcobalamin Promotes the Differentiation of Schwann Cells and Remyelination in Lysophosphatidylcholine-Induced Demyelination of the Rat Sciatic Nerve. Front. Cell. Neurosci. 2015, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Gröber, U.; Kisters, K.; Schmidt, J. Neuroenhancement with Vitamin B12-Underestimated Neurological Significance. Nutrients 2013, 5, 5031–5045. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Models | Induction of VitB12 Deficiency | Treatment | Reference |
|---|---|---|---|
| Mouse | Cbi supplementation through osmotic minipumps | 21 female mice, 27 days treatment | [48] |
| |||
| |||
| |||
| Mouse | EtPhCbl supplementation through osmotic minipumps | 18 female mice, 27 days treatment | [49] |
| |||
| |||
| |||
| Mouse | RoninF80L/F80L and Hcfc1A115V/Y mouse model using CRISPR/Cas9 genome editing | / | [50] |
| Mouse | 1. Mmachcflox/flox mouse model generated using CRISPR/Cas9 genome editing | / | [37] |
| 2. Mmachc-OE+/tg transgenic mouse line that over-expresses functional mmachc | |||
| Rat | Rats made VitB12 deficient by total gastrectomy | / | [51] |
| Rat | Rats administered with N2O (which induces the irreversible oxidation of Co+ to the Co+++ form, rendering VitB12 inactive). | / | [52] |
| Zebrafish | Embryos carrying the c.95_132delins28 p.Gly32Valfs*48 mutation in the mmachc (hg13) gene in a homozygous state | / | [53] |
| Zebrafish | 1. mmachc morphant mutant created using morpholinos or simRNA to target the mRNA of embryos | / | [54] |
| 2. Germinal mutant created using zinc finger nucleases (ZFNs) approach |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathew, A.R.; Di Matteo, G.; La Rosa, P.; Barbati, S.A.; Mannina, L.; Moreno, S.; Tata, A.M.; Cavallucci, V.; Fidaleo, M. Vitamin B12 Deficiency and the Nervous System: Beyond Metabolic Decompensation—Comparing Biological Models and Gaining New Insights into Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2024, 25, 590. https://doi.org/10.3390/ijms25010590
Mathew AR, Di Matteo G, La Rosa P, Barbati SA, Mannina L, Moreno S, Tata AM, Cavallucci V, Fidaleo M. Vitamin B12 Deficiency and the Nervous System: Beyond Metabolic Decompensation—Comparing Biological Models and Gaining New Insights into Molecular and Cellular Mechanisms. International Journal of Molecular Sciences. 2024; 25(1):590. https://doi.org/10.3390/ijms25010590
Chicago/Turabian StyleMathew, Aimee Rachel, Giacomo Di Matteo, Piergiorgio La Rosa, Saviana Antonella Barbati, Luisa Mannina, Sandra Moreno, Ada Maria Tata, Virve Cavallucci, and Marco Fidaleo. 2024. "Vitamin B12 Deficiency and the Nervous System: Beyond Metabolic Decompensation—Comparing Biological Models and Gaining New Insights into Molecular and Cellular Mechanisms" International Journal of Molecular Sciences 25, no. 1: 590. https://doi.org/10.3390/ijms25010590