
New Brassinosteroid Analogs with 23,24-Dinorcholan Side Chain, and Benzoate Function at C-22: Synthesis, Assessment of Bioactivity on Plant Growth, and Molecular Docking Study

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
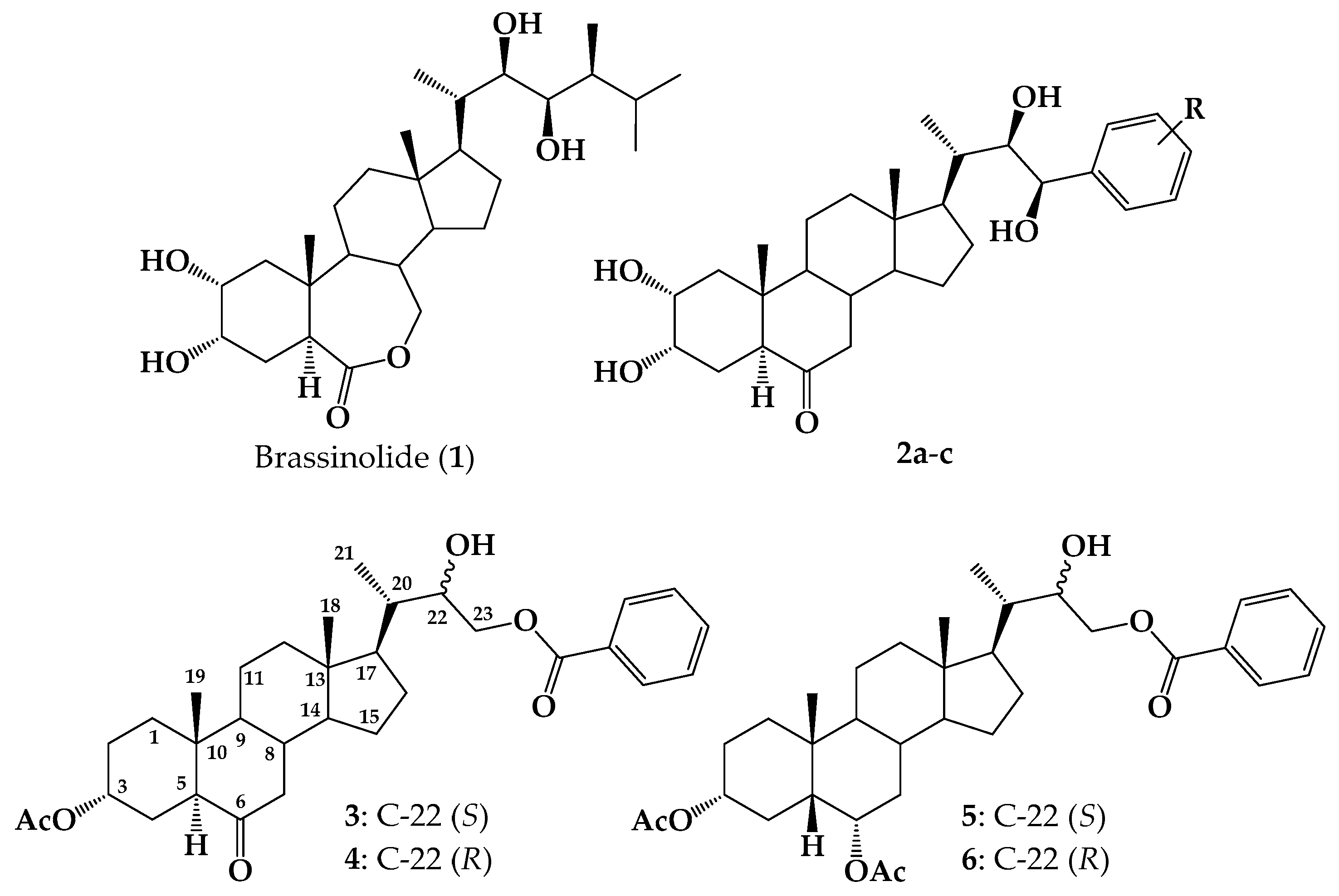
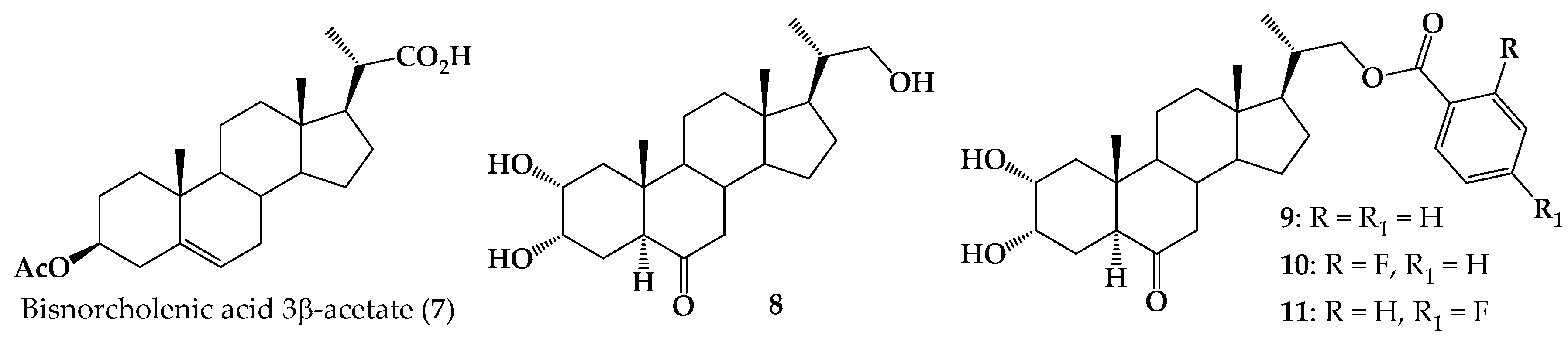
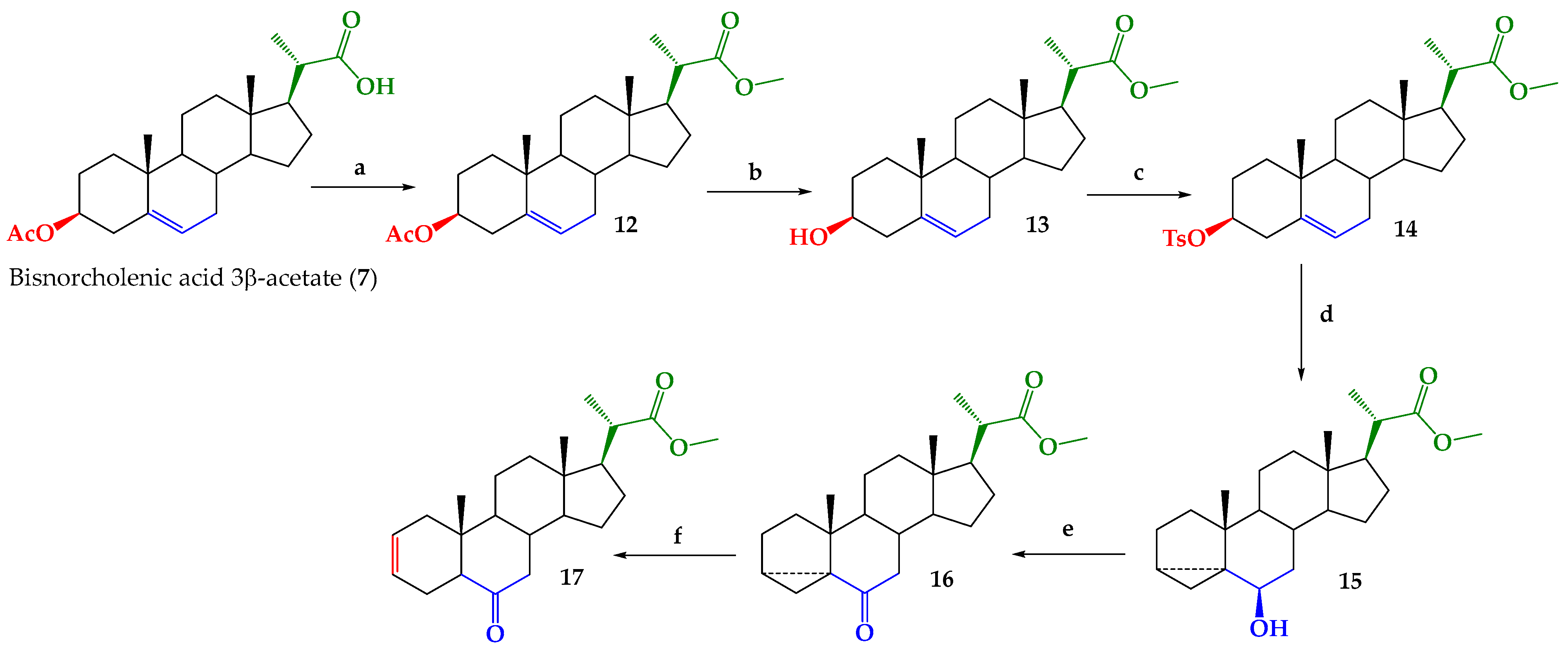
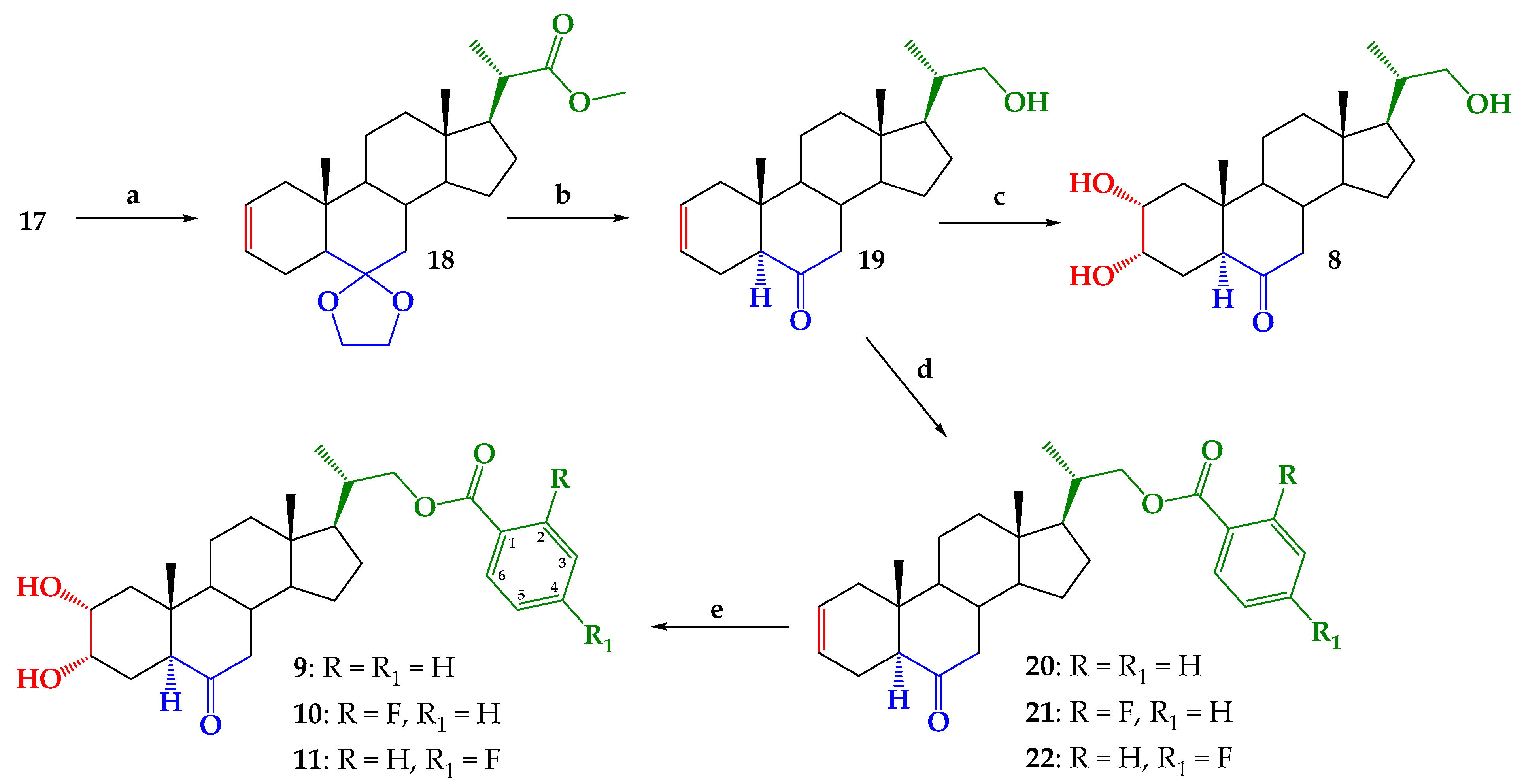
2.1. Chemical Synthesis
2.2. Biological Activity
2.2.1. Bioactivity in the Rice Lamina Inclination Test (RLIT) of Brassinosteroid Analogs
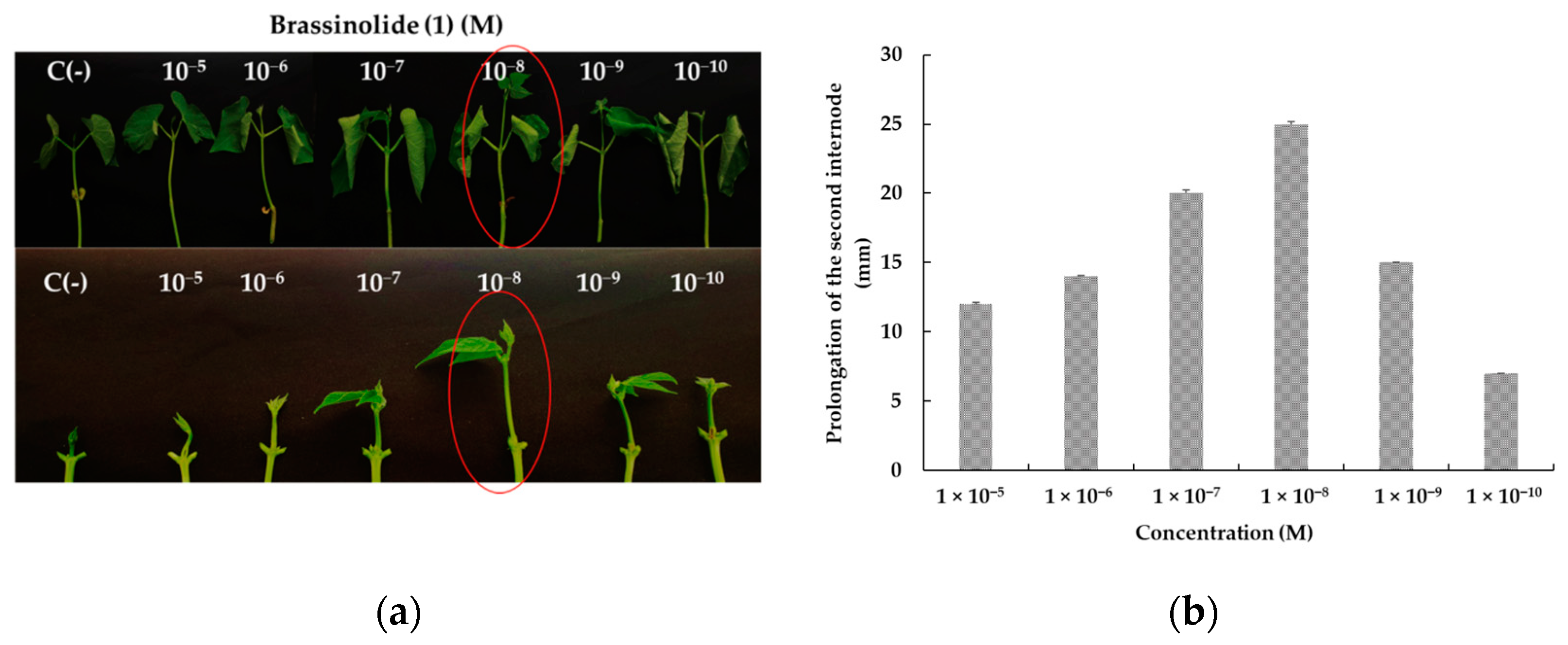
2.2.2. Bioactivity in Bean Second Internode Bioassay of Brassinosteroid Analogs
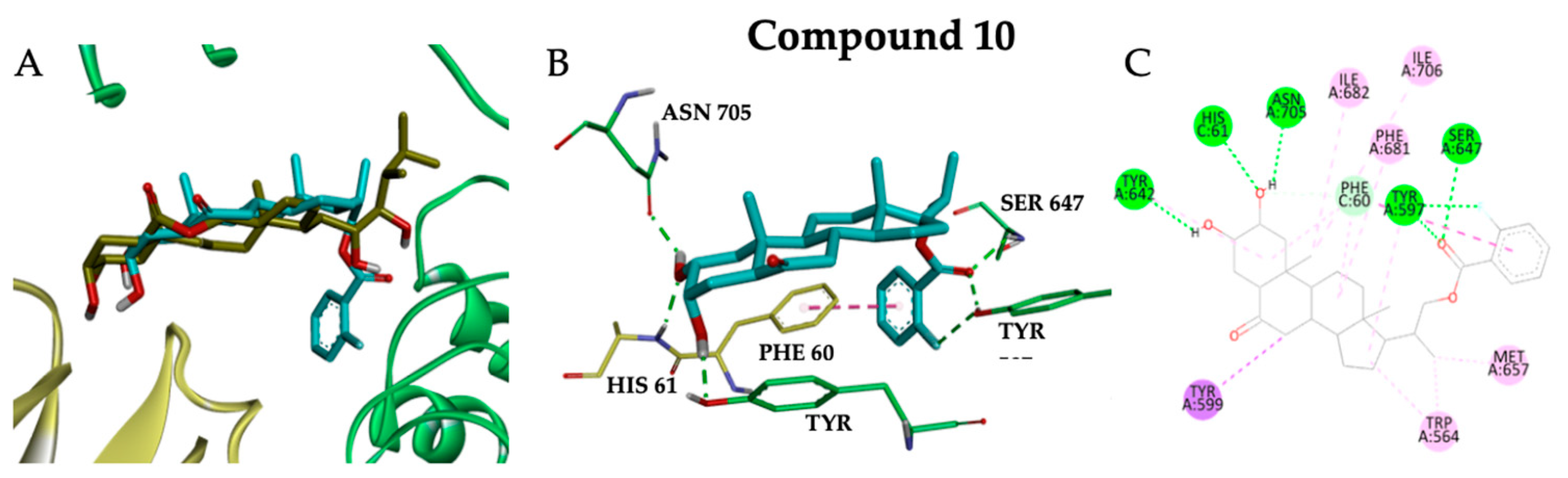
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. General Chemical
3.2. Synthesis
3.2.1. Methyl (20S)-3β-Acetoxypregn-5-ene-20-carboxylate (12)
3.2.2. Methyl (20S)-3β-Hydroxy-pregn-5-ene-20-carboxylate (13)
3.2.3. Methyl (20S)-3β-(4-Toluennsulfonyloxy)-pregn-5-ene-20-carboxylate (14)
3.2.4. Methyl (20S)-6β-Hydroxy-3α,5-cyclo-5α-pregnane-20-carboxylate (15)
3.2.5. Methyl (20S)-6-Oxo-3α,5-cyclo-5α-pregnane-20-carboxylate (16)
3.2.6. Methyl (20S)-6-Oxo-5α-pregn-2-ene-20-carboxylate (17)
3.2.7. Methyl (20S)-6,6-Ethylenedioxy-5α-pregn-2-ene-20-carboxylate (18)
3.2.8. 22-Hydroxy-5α-cholan-2-ene-23,24-dinor-6-one (19)
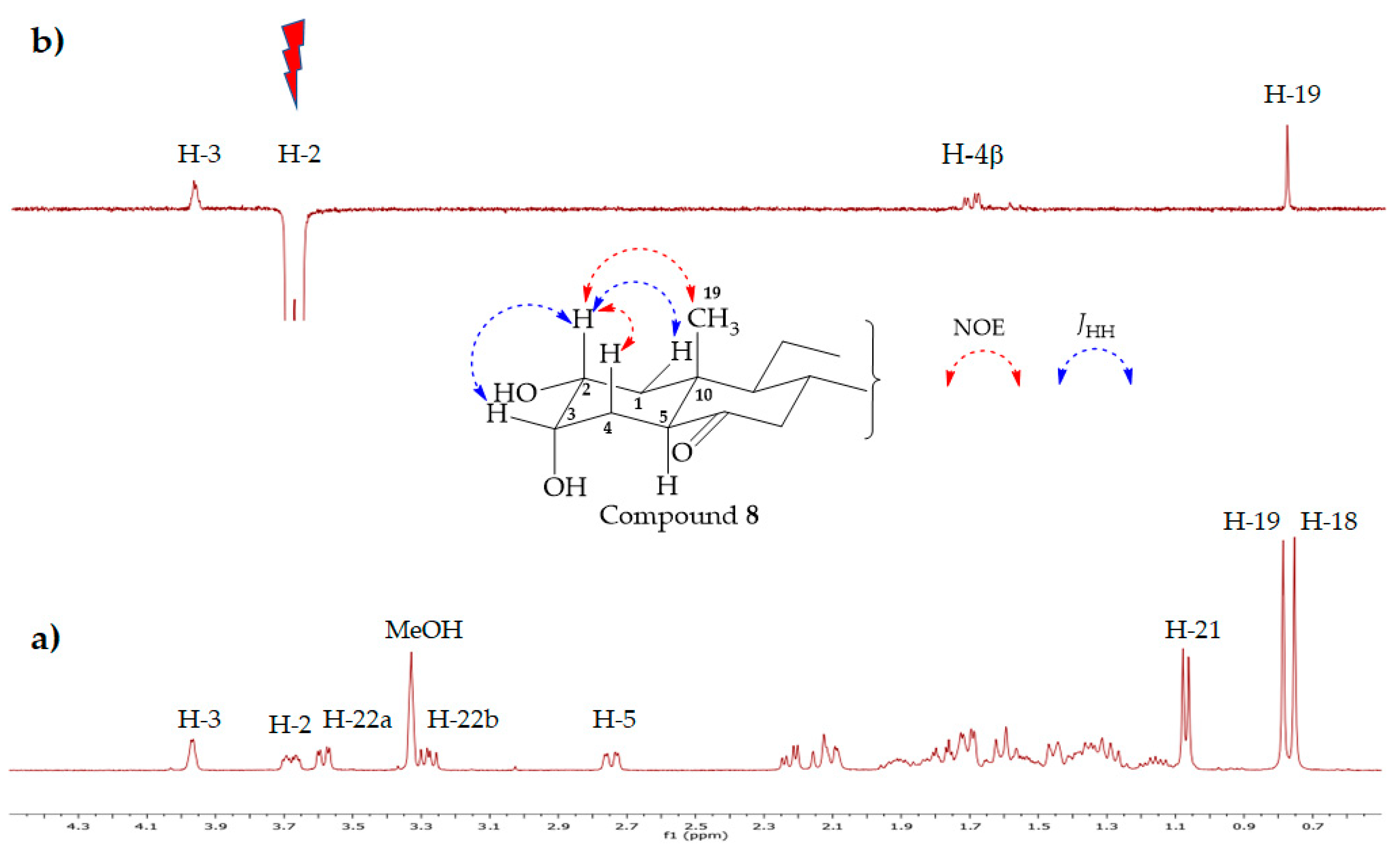
3.2.9. 2α,3α,22-Thrihydroxy-5α-cholan-23,24-dinor-6-one (8)
3.2.10. General Procedure for Synthesis of 5α-Cholan-6-oxo-2-ene-23,24-dinor-22-(substituted)-benzoate-22-yl (20–22)
3.2.11. 5α-Cholan-6-oxo-2-ene-23,24-dinor-22-benzoate-22-yl (20)
3.2.12. 5α-Cholan-6-oxo-2-ene-23,24-dinor-22-(2-Fluoro) benzoate-22-yl (21)
3.2.13. 5α-Cholan-6-oxo-2-ene-23,24-dinor-22-(4-Fluoro)-benzoate-22-yl (22)
3.2.14. General Procedure for Synthesis of 2α,3α-Dihydroxy-5α-cholan-6-oxo-23,24-dinor-22-(4-substituted)-benzoate-22-yl (9–11) by Sharpless Dihydroxylation [37,50]
3.2.15. 2α,3α-Dihydroxy-5α-cholan-6-oxo-23,24-dinor-22-benzoate-22-yl (9)
3.2.16. 2α,3α-Dihydroxy-5α-cholan-6-oxo-23,24-dinor-22-(2-Fluoro)-benzoate-22-yl (10)
3.2.17. 2α,3α-Dihydroxy-5α-cholan-6-oxo-23,24-dinor-22-(4-Fluoro)-benzoate-22-yl (11)
3.3. Biological Activity
3.3.1. Bioactivity in the Rice Lamina Inclination Test (RLIT) of Brassinosteroids Analogs
3.3.2. Bean Second Internode Bioassay
3.4. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujioka, S. Natural Occurrence of Brassinosteroids in the Plant Kingdom. In Brassinosteroids: Steroidal Plant Hormones; Sakurai, A., Yokota, T., Clouse, S.D., Eds.; Springer: Tokyo, Japan, 1999; pp. 21–45. [Google Scholar]
- Bajguz, A. Brassinosteroids—Occurence and chemical structures in plants. In Brassinosteroids: A Class of Plant Hormone; Hayat, S., Ahmad, A., Eds.; Springer: Dordrecht, The Netherlands, 2016; pp. 1–27. [Google Scholar]
- Clouse, S.D.; Sasse, J.M. Brassinosteroids: Essential regulators of plant growth and development. Annu. Rev. Plant Biol. 1998, 49, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.W.; Mandava, N.; Worley, J.F.; Plimmer, J.R.; Smith, M.V. Brassins—A New Family of Plant Hormones from Rape Pollen. Nature 1970, 225, 1065–1066. [Google Scholar] [CrossRef] [PubMed]
- Oklestkova, J.; Rarova, L.; Kvasnica, M.; Strnad, M. Brassinosteroids: Synthesis and biological activities. Phytochem. Rev. 2015, 14, 1053–1072. [Google Scholar] [CrossRef]
- Clouse, S.D. A History of Brassinosteroid Research from 1970 through 2005: Thirty-Five Years of Phytochemistry, Physiology, Genes, and Mutants. J. Plant Growth Regul. 2015, 34, 828–844. [Google Scholar] [CrossRef]
- Nolan, T.M.; Vukašinović, N.; Liu, D.; Russinova, E.; Yin, Y. Brassinosteroids: Multidimensional Regulators of Plant Growth, Development, and Stress Responses. Plant Cell 2020, 32, 295. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zheng, H.; Lin, L.; Wang, F.; Sui, N. Roles of brassinosteroids in plant growth and abiotic stress response. Plant Growth Regul. 2021, 93, 29–38. [Google Scholar] [CrossRef]
- Hafeez, M.B.; Zahra, N.; Zahra, K.; Raza, A.; Batool, A.; Shaukat, K.; Khan, S. Brassinosteroids: Molecular and physiological responses in plant growth and abiotic stresses. Plant Stress 2021, 2, 100029. [Google Scholar] [CrossRef]
- Khripach, V.A.; Zhabinskii, V.; de Groot, A.E. Brassinosteroids: A New Class of Plant Hormones; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Herrera, H.; Carvajal, R.; Olea, A.F.; Espinoza, L. Structural modifications of deoxycholic acid to obtain three known brassinosteroid analogues and full NMR spectroscopic characterization. Molecules 2016, 21, 1139. [Google Scholar] [CrossRef]
- Tian, W.S.; Zhou, W.S.; Jiang, B.; Pan, X.F. Studies on Steroidal Plant-Growth Regulator 9. The Preparation of 22R-Penta-Nor-Brassinolides and 22S-24,25,26,27,28-Penta-Nor-Brassinolides. Acta Chim. Sin. 1989, 47, 1017–1021. [Google Scholar]
- Espinoza, L.; Cortes, M. Synthesis and biological activities of two new brassinosteroids functionalized in ring C. Bol. Soc. Chil. Quim. 2002, 47, 335–347. [Google Scholar]
- Espinoza, L.; Cortes, M. Synthesis and biological activity of brassinosteroids analogues. Bol. Soc. Chil. Quim. 2002, 47, 511–516. [Google Scholar]
- Espinoza, L. Synthesis of Four New Brassinosteroids Analogues 11-Oxo-Functionalized on C Ring, with 24-Nor Side Chain and Containing 5â-Cholanic Acid Skeleton. Org. Chem. Curr. Res. 2015, 4, 1–6. [Google Scholar]
- Carvajal, R.; Gonzalez, C.; Olea, A.F.; Fuentealba, M.; Espinoza, L. Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid. Molecules 2018, 23, 1306. [Google Scholar] [CrossRef] [PubMed]
- Diaz, K.; Espinoza, L.; Carvajal, R.; Conde-Gonzalez, M.; Niebla, V.; Olea, A.F.; Coll, Y. Biological Activities and Molecular Docking of Brassinosteroids 24-Norcholane Type Analogs. Int. J. Mol. Sci. 2020, 21, 1832. [Google Scholar] [CrossRef] [PubMed]
- Soto, N.; González, C.; Mellado, M.; Olea, A.F.; Coll, Y.; Díaz, K.; Espinoza, L. Epimeric Mixtures of Brassinosteroid Analogs: Synthesis, Plant Growth, and Germination Effects in Tomato (Lycopersicum esculentum Mill.). Agronomy 2020, 10, 808. [Google Scholar] [CrossRef]
- Voigt, B.; Porzel, A.; Golsch, D.; Adam, W.; Adam, G. Regioselective oxyfunctionalization of brassinosteroids by methyl(trifluoromethyl)dioxirane: Synthesis of 25-hydroxy-brassinolide and 25-hydroxy-24-epibrassinolide by direct C-H insertion. Tetrahedron 1996, 52, 10653–10658. [Google Scholar] [CrossRef]
- Brosa, C.; Capdevila, J.M.; Zamora, I. Brassinosteroids: A new way to define the structural requirements. Tetrahedron 1996, 52, 2435–2448. [Google Scholar] [CrossRef]
- Voigt, B.; Porzel, A.; Bruhn, C.; Wagner, C.; Merzweiler, K.; Adam, G. Synthesis of 24-epicathasterone and related brassinosteroids with modified side chain. Tetrahedron 1997, 53, 17039–17054. [Google Scholar] [CrossRef]
- Seto, H.; Fujioka, S.; Koshino, H.; Yoshida, S.; Tsubuki, M.; Honda, T. Synthesis and biological evaluation of extra-hydroxylated brassinolide analogs. Tetrahedron 1999, 55, 8341–8352. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.; Gil, R.P.; Leliebre-Lara, V.; Martinez, C.S.P.; Manchado, F. Synthesis and biological activity of (22R,25R)-5α-furostan-2α,3α,26-triol. J. Chem. Res. 1996, 504–505. [Google Scholar]
- Iglesias-Arteaga, M.; Gil, R.; Leliebre-Lara, V.; Coll-Manchado, F.; Pérez, C.S.; Rosado, A. Synthesis of (25R)-5α-Spirostan-2α,3α,6β-triol Triacetate. Synth. Commun. 1998, 28, 75–81. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.; Gil, R.P.; Leliebre-Lara, V.; Martinez, C.S.P.; Manchado, F.; Perez, A.R.; Rios, L.P. Synthesis of (22R,25R)-3β,26-dihydroxy-5α-furostan-6-one. Synth. Commun. 1998, 28, 1381–1386. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.; Gil, R.P.; Leliebre-Lara, V.; Martinez, C.S.P.; Manchado, F. Synthesis of (22R,25R)-2α,3α,26-trihydroxy-5α-furostanaone-6-one. Synth. Commun. 1998, 28, 1779–1784. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; PérezGil, R.; LeliebreLara, V.; CollManchado, F.; PérezMartínez, C.S. Synthesis of (25R)-2α,3α-Epoxy-5α-Spirostan-6,23-Dione. Synth. Commun. 1998, 28, 4387–4392. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; Martinez, C.S.P.; Manchado, F.C. Synthesis and characterization of (25R)-2α,3α-epoxy-5α-spirostan-12,23-dione. Synth. Commun. 1999, 29, 1811–1818. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; Gil, R.-P.; Pérez-Martínez, C.S.; Coll-Manchado, F. Synthetic Steroidal Sapogenins. Part III1 23-Ketohecogenin and 23-Ketoisochiapagenin. Synth. Commun. 2000, 30, 163–170. [Google Scholar] [CrossRef]
- Back, T.; Pharis, R. Structure-Activity Studies of Brassinosteroids and the Search for Novel Analogues and Mimetics with Improved Bioactivity. J. Plant Growth Regul. 2004, 22, 350–361. [Google Scholar] [CrossRef]
- Zhou, W.S.; Tian, W.S. The Synthesis of Steroids Containing Structural Unit of A, B Ring of Brassinolide and Ecdysone from Hyodeoxycholic Acid. Acta Chim. Sin. 1984, 42, 1173–1177. [Google Scholar]
- Zhou, W.; Jiang, B.; Shen, J. Synthesis of Cholesteric Lactones and Analogs as Plant Growth Regulators. Patent CN 1184113 A, 10 June 1998. [Google Scholar]
- Duran, M.I.; Gonzalez, C.; Acosta, A.; Olea, A.F.; Diaz, K.; Espinoza, L. Synthesis of Five Known Brassinosteroid Analogs from Hyodeoxycholic Acid and Their Activities as Plant-Growth Regulators. Int. J. Mol. Sci. 2017, 18, 516. [Google Scholar] [CrossRef] [PubMed]
- Diachkov, M.V.; Ferrer, K.; Oklestkova, J.; Rarova, L.; Bazgier, V.; Kvasnica, M. Synthesis and Biological Activity of Brassinosteroid Analogues with a Nitrogen-Containing Side Chain. Int. J. Mol. Sci. 2021, 22, 155. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.F.; Zhou, W.S. Studies on Steroidal Plant-Growth Regulators. Part 33. Novel Method for Construction of the Side-Chain of 23-Arylbrassinosteroids Via Heck Arylation and Asymmetric Dihydroxylation As Key Steps. J. Chem. Soc. Perkin Trans. 1 1994, 24, 3579–3585. [Google Scholar] [CrossRef]
- Cimino, F.P.; Núñez, M.G.; Rosado-Abón, A.; Amesty, Á.; Estévez-Braun, A.; Díaz, K.; Espinoza, L.C.; Iglesias-Arteaga, M.A. Methyl esters of 23,24-Dinor-5α-cholan-22-oic acids as brassinosteroid Analogues. Synthesis, evaluation of plant growth promoting activity and Molecular docking. Steroids 2023, 196, 109248. [Google Scholar] [CrossRef]
- Kvasnica, M.; Oklestkova, J.; Bazgier, V.; Rárová, L.; Korinkova, P.; Mikulík, J.; Budesinsky, M.; Béres, T.; Berka, K.; Lu, Q.; et al. Design, synthesis and biological activities of new brassinosteroid analogues with a phenyl group in the side chain. Org. Biomol. Chem. 2016, 14, 8691–8701. [Google Scholar] [CrossRef]
- Korinkova, P.; Bazgier, V.; Oklestkova, J.; Rarova, L.; Strnad, M.; Kvasnica, M. Synthesis of novel aryl brassinosteroids through alkene cross-metathesis and preliminary biological study. Steroids 2017, 127, 46–55. [Google Scholar] [CrossRef]
- Ferrer, K.; Díaz, K.; Kvasnica, M.; Olea, A.F.; Cuellar, M.; Espinoza, L. Synthesis of New Brassinosteroid 24-Norcholane Type Analogs Conjugated in C-3 with Benzoate Groups. Molecules 2021, 26, 1173. [Google Scholar] [CrossRef]
- Soto, N.; Ferrer, K.; Díaz, K.; González, C.; Taborga, L.; Olea, A.F.; Carrasco, H.; Espinoza, L. Synthesis and Biological Activity of New Brassinosteroid Analogs of Type 24-Nor-5β-Cholane and 23-Benzoate Function in the Side Chain. Int. J. Mol. Sci. 2021, 22, 4808. [Google Scholar] [CrossRef]
- She, J.; Han, Z.; Kim, T.-W.; Wang, J.; Cheng, W.; Chang, J.; Shi, S.; Wang, J.; Yang, M.; Wang, Z.-Y.; et al. Structural insight into brassinosteroid perception by BRI1. Nature 2011, 474, 472. [Google Scholar] [CrossRef]
- Hothorn, M.; Belkhadir, Y.; Dreux, M.; Dabi, T.; Noel, J.P.; Wilson, I.A.; Chory, J. Structural basis of steroid hormone perception by the receptor kinase BRI1. Nature 2011, 474, 467–471. [Google Scholar] [CrossRef]
- Nam, K.H.; Li, J. BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Han, Z.; Tang, J.; Hu, Z.; Chai, C.; Zhou, B.; Chai, J. Structure reveals that BAK1 as a co-receptor recognizes the BRI1-bound brassinolide. Cell Res. 2013, 23, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Han, Z.; Zhou, B.; Chai, J. Structural basis for differential recognition of brassinolide by its receptors. Protein Cell 2013, 4, 475–482. [Google Scholar] [CrossRef]
- Černý, V.; Strnad, M.; Kamínek, M. Preparation of 2α,3α-dihydroxy-7-oxa-6-oxo-23,24-dinor-B-homo-5α-cholanic acid, its esters and amides as brassinolide analogues. Collect. Czech. Chem. Commun. 1986, 51, 687–697. [Google Scholar] [CrossRef]
- Kohout, L.J.; Chodounská, H.; Macek, T.; Strnad, M. Synthesis of (20S)-2α,3α-Dihydroxy-6-oxo-7-oxa-7a-homo-5α-pregnane-20-carboxylic Acid as a Brassinosteroid Part of Ligands for Binding to Affinity Chromatography Carriers. Collect. Czech. Chem. Commun. 2000, 65, 1754–1761. [Google Scholar] [CrossRef]
- Antonchick, A.P.; Schneider, B.; Zhabinskii, V.N.; Khripach, V.A. Synthesis of [26,27-2H6] brassinosteroids from 23,24-bisnorcholenic acid methyl ester. Steroids 2004, 69, 617–628. [Google Scholar] [CrossRef]
- Kondo, M.; Mori, K. Synthesis of Brassinolide Analogs with or without the Steroidal Side Chain. Agric. Biol. Chem. 1983, 47, 97–102. [Google Scholar] [CrossRef]
- Oyarce, J.; Aitken, V.; Gonzalez, C.; Ferrer, K.; Olea, A.F.; Parella, T.; Espinoza, L. Synthesis and structural determination of new brassinosteroid 24-nor-5α-cholane type analogs. Molecules 2019, 24, 4612. [Google Scholar] [CrossRef]
- Oklestkova, J.; Rárová, L.; Strnad, M. Brassinosteroids and their Biological Activities. In Natural Products; Ramawat, K., Merillon, J., Eds.; Springer: Berlin/Heilderberg, Germany, 2013; pp. 3851–3871. [Google Scholar]
- Zullo, M.A.T.; Adam, G. Brassinosteroid phytohormones: Structure, bioactivity and applications. Braz. J. Plant Physiol. 2002, 14, 143–181. [Google Scholar] [CrossRef]
- Kvasnica, M.; Oklestkova, J.; Bazgier, V.; Rarova, L.; Berka, K.; Strnad, M. Biological activities of new monohydroxylated brassinosteroid analogues with a carboxylic group in the side chain. Steroids 2014, 85, 58–64. [Google Scholar] [CrossRef]
- Kerb, U.; Eder, U.; Krähmer, H. Synthesis of Hexanor-Brassinolide-22-ethers with Plant Growth-promoting Activity. Agric. Biol. Chem. 1986, 50, 1359–1360. [Google Scholar]
- Eignerová, B.; Slavíková, B.; Buděšínský, M.; Dračínský, M.; Klepetářová, B.; Št’astná, E.; Kotora, M. Synthesis of Fluorinated Brassinosteroids Based on Alkene Cross-Metathesis and Preliminary Biological Assessment. J. Med. Chem. 2009, 52, 5753–5757. [Google Scholar] [CrossRef] [PubMed]
- Kohout, L.; Strnad, M. Brassinosteroids with ester function with five carbon atoms at the 20 position. Collect. Czech. Chem. Commun. 1992, 57, 1731–1738. [Google Scholar] [CrossRef]
- Rosado-Abon, A.; de Dios-Bravo, G.; Rodriguez-Sotres, R.; Iglesias-Arteaga, M.A. Synthesis and plant growth promoting activity of polyhydroxylated ketones bearing the 5 alpha-hydroxy-6-oxo moiety and cholestane side chain. Steroids 2012, 77, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Rosado-Abón, A.; de Dios-Bravo, G.; Rodríguez-Sotres, R.; Iglesias-Arteaga, M.A. Synthesis and plant growth promoting activity of dinorcholanic lactones bearing the 5α-hydroxy-6-oxo moiety. J. Steroid Biochem. Mol. Biol. 2013, 134, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.; Jang, S. Rice Lamina Joint Inclination Assay. Bio-Protocol 2017, 7, e2409. [Google Scholar] [CrossRef] [PubMed]
- Slavikova, B.; Kohout, L.; Budesinsky, M.; Swaczynova, J.; Kasal, A. Brassinosteroids: Synthesis and Activity of Some Fluoro Analogues. J. Med. Chem. 2008, 51, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA; Discovery Studio Modeling Environment, Release 2021; Dassault Systèmes: San Diego, CA, USA, 2021.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bending Angle between Lamina and Sheaths (Degrees ± Standard Error) 1 | ||
|---|---|---|
| Compounds | 1 × 10−7 M | 1 × 10−6 M |
| 1 | 46 ± 8.0 a | 89 ± 4.9 a |
| 8 | 26 ± 6.1 c | 16 ± 4.1 d |
| 9 | 21 ± 2.0 c | 10 ± 2.6 e |
| 10 | 42 ± 0.0 a | 24 ± 2.7 c |
| 11 | 33 ± 2.0 b | 36 ± 2.0 b |
| Compounds | Elongation of the Second Internode, mm ± SD |
|---|---|
| 1 | 20.2 ± 0.65 b |
| 8 | 8.5 ± 0.49 c |
| 9 | 20.8 ± 0.26 b |
| 10 | 17.5 ± 0.52 b |
| 11 | 85.0 ± 4.00 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, V.; Diaz, K.; Soto, M.; Olea, A.F.; Cuellar, M.A.; Nuñez, M.; Espinoza-Catalán, L. New Brassinosteroid Analogs with 23,24-Dinorcholan Side Chain, and Benzoate Function at C-22: Synthesis, Assessment of Bioactivity on Plant Growth, and Molecular Docking Study. Int. J. Mol. Sci. 2024, 25, 419. https://doi.org/10.3390/ijms25010419
Aitken V, Diaz K, Soto M, Olea AF, Cuellar MA, Nuñez M, Espinoza-Catalán L. New Brassinosteroid Analogs with 23,24-Dinorcholan Side Chain, and Benzoate Function at C-22: Synthesis, Assessment of Bioactivity on Plant Growth, and Molecular Docking Study. International Journal of Molecular Sciences. 2024; 25(1):419. https://doi.org/10.3390/ijms25010419
Chicago/Turabian StyleAitken, Vanessa, Katy Diaz, Mauricio Soto, Andrés F. Olea, Mauricio A. Cuellar, Maria Nuñez, and Luis Espinoza-Catalán. 2024. "New Brassinosteroid Analogs with 23,24-Dinorcholan Side Chain, and Benzoate Function at C-22: Synthesis, Assessment of Bioactivity on Plant Growth, and Molecular Docking Study" International Journal of Molecular Sciences 25, no. 1: 419. https://doi.org/10.3390/ijms25010419