
Advantages of Induced Circular Dichroism Spectroscopy for Qualitative and Quantitative Analysis of Solution-Phase Cyclodextrin Host–Guest Complexes

Abstract
:1. Introduction
2. Results
2.1. CD and UV Spectroscopy Measurements
2.1.1. Comparison of Bifonazole and Fenbufen
2.1.2. Comparison of Fenoprofen and Nimesulide
2.2. NMR Measurements
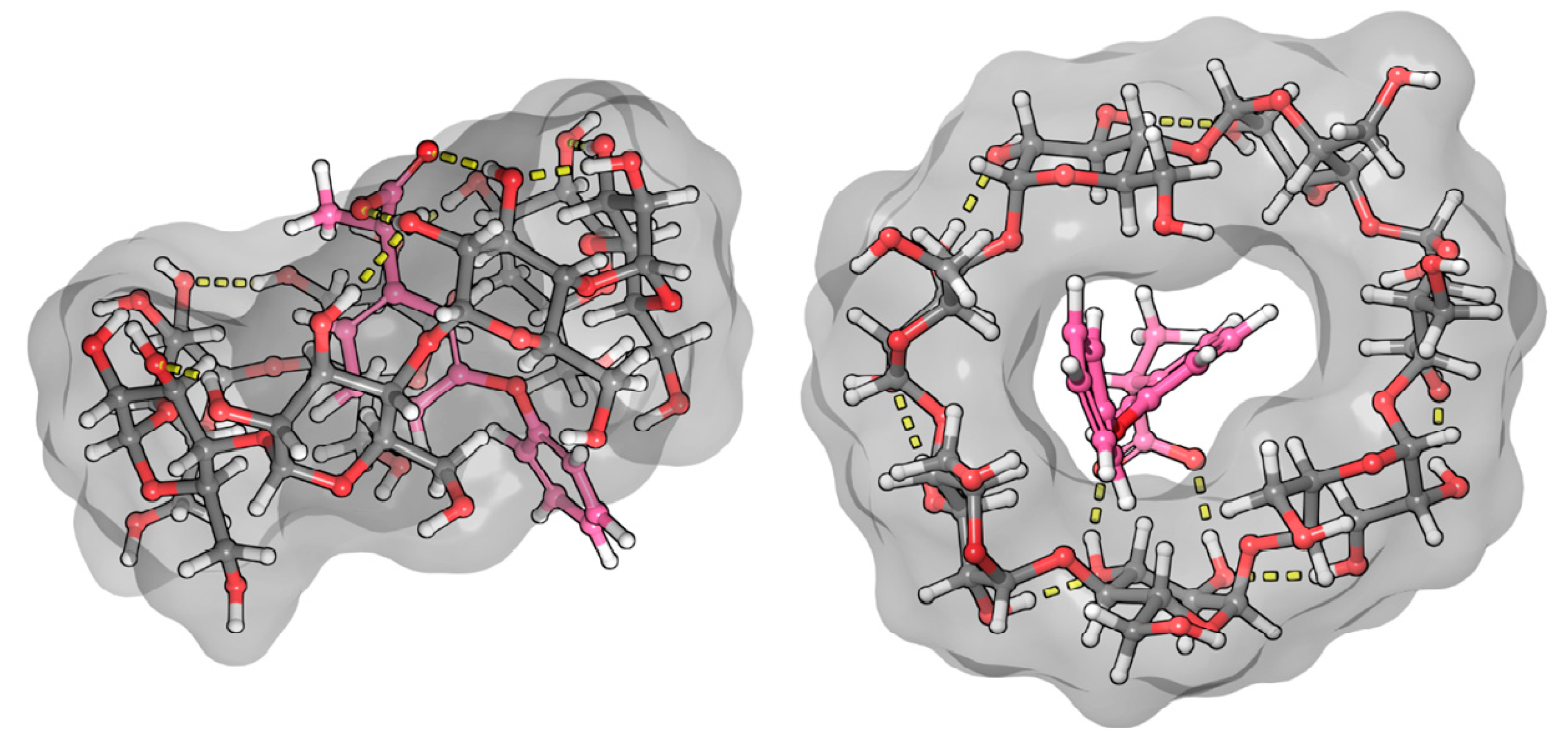
2.3. Molecular Dynamic and Quantum Chemical Calculations
3. Discussion
3.1. Bifonazole and Fenbufen
3.1.1. Bifonazole
3.1.2. Fenbufen
3.2. Fenoprofen and Nimesulide
3.2.1. Fenoprofen
3.2.2. Nimesulide
3.3. Transition Dipole Moment Vectors
4. Materials and Methods
4.1. Materials
4.2. NMR Measurements
4.3. CD Measurements
Preparation of Solutions for CD Measurements
4.4. Computational Methods
4.4.1. Preparation of the Compounds
4.4.2. Preparation of β-Cyclodextrin
4.4.3. Molecule Dynamic Simulations
4.4.4. Quantum Mechanics Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FDA Developing Products for Rare Diseases & Conditions: Hydroxy-Propyl-Beta-Cyclodextrin. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=303910 (accessed on 19 November 2023).
- Cyclodextrins used as excipients (EMA/CHMP/495747/2013). Available online: https://www.ema.europa.eu/en/cyclodextrins (accessed on 19 November 2023).
- Puskás, I.; Szente, L.; Szőcs, L.; Fenyvesi, É. Recent List of Cyclodextrin-Containing Drug Products. Period. Polytech. Chem. Eng. 2023, 67, 11–17. [Google Scholar] [CrossRef]
- Sarabia-Vallejo, Á.; Caja, M.d.M.; Olives, A.I.; Martín, M.A.; Menéndez, J.C. Cyclodextrin Inclusion Complexes for Improved Drug Bioavailability and Activity: Synthetic and Analytical Aspects. Pharmaceutics 2023, 15, 2345. [Google Scholar] [CrossRef] [PubMed]
- Venuti, V.; Crupi, V.; Fazio, B.; Majolino, D.; Acri, G.; Testagrossa, B.; Stancanelli, R.; De Gaetano, F.; Gagliardi, A.; Paolino, D.; et al. Physicochemical Characterization and Antioxidant Activity Evaluation of Idebenone/Hydroxypropyl-β-Cyclodextrin Inclusion Complex. Biomolecules 2019, 9, 531. [Google Scholar] [CrossRef] [PubMed]
- Szente, L.; Puskás, I.; Sohajda, T.; Varga, E.; Vass, P.; Nagy, Z.K.; Farkas, A.; Várnai, B.; Béni, S.; Hazai, E. Sulfobutylether-Beta-Cyclodextrin-Enabled Antiviral Remdesivir: Characterization of Electrospun- and Lyophilized Formulations. Carbohydr. Polym. 2021, 264, 118011. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, A.H.; Szeleszczuk, Ł. A Review of Applications of Solid-State Nuclear Magnetic Resonance (ssNMR) for the Analysis of Cyclodextrin-Including Systems. Int. J. Mol. Sci. 2023, 24, 3648. [Google Scholar] [CrossRef] [PubMed]
- Hădărugă, N.G.; Bandur, G.N.; David, I.; Hădărugă, D.I. A review on thermal analyses of cyclodextrins and cyclodextrin complexes. Environ. Chem. Lett. 2019, 17, 349–373. [Google Scholar] [CrossRef]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in the solid state: A review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef]
- Hans-Jörg, S.; Hacket, F.; Rüdiger, V.; Ikeda, H. NMR studies of cyclodextrines and cyclodextrin complexes. Chem. Rev. 1998, 5, 1755–1786. [Google Scholar] [CrossRef]
- Haouas, M.; Falaise, C.; Leclerc, N.; Floquet, S.; Cadot, E. NMR spectroscopy to study cyclodextrin-based host–guest assemblies with polynuclear clusters. Dalton Trans. 2023, 52, 13467–13481. [Google Scholar] [CrossRef]
- Granadero, D.; Bordello, J.; Pérez-Alvite, M.J.; Novo, M.; Al-Soufi, W. Host-Guest Complexation Studied by Fluorescence Correlation Spectroscopy: Adamantane–Cyclodextrin Inclusion. Int. J. Mol. Sci. 2010, 11, 173–188. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, W.; Lv, H.; Tong, S. Spectral study on inclusion interaction and enantiorecognition of 2-aryl carboxylic acids with hydroxypropyl-β-cyclodextrin. Chirality 2020, 32, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Hou, J.; Hu, J.; Geng, Y.; Li, M.; Wang, H.; Wang, J.; Luo, Q. Recent advances in β-cyclodextrin-based materials for chiral recognition. Chem. Commun. 2023, 60, 9157–9166. [Google Scholar] [CrossRef] [PubMed]
- Chankvetadze, B.; Scriba, G.K.E. Cyclodextrins as chiral selectors in capillary electrophoresis: Recent trends in mechanistic studies. TrAC Trends Anal. Chem. 2023, 160, 116987–117001. [Google Scholar] [CrossRef]
- El Deeb, S.; Silva, C.F.; Junior, C.S.N.; Hanafi, R.S.; Borges, K.B. Chiral Capillary Electrokinetic Chromatography: Principle and Applications, Detection and Identification, Design of Experiment, and Exploration of Chiral Recognition Using Molecular Modeling. Molecules 2021, 26, 2841. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, M.L.; Galarreta, B.C.; Mahon, P.J.; McCurry, D.A.; Gerdon, A.E.; Collier, S.M.; Squires, M.E. Survey of the undergraduate analytical chemistry curriculum. J. Chem. Educ. 2022, 99, 2317–2326. [Google Scholar] [CrossRef]
- Dodziuk, H.; Danikiewicz, W.; Grabner, G.; Krois, D.; Brinker, U.H.; Bilewicz, R.; Chmurski, K.; Kunitake, M.; Ohira, A. Circular Dichroism of Cyclodextrin Complexes. In Cyclodextrins and Their Complexes; Dodziuk, H., Ed.; Wiley Online Books; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Berova, N.; Polavarapu, P.L.; Nakanishi, K.; Woody, R.W. Comprehensive Chiroptical Spectroscopy Vol I: Instrumentation, Methodologies and Theoretical Simulations; Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Gawroński, J.; Grajewski, J. The significance of induced circular dichroism. Org. Lett. 2003, 5, 3301–3303. [Google Scholar] [CrossRef] [PubMed]
- Kiss, E.; Szabó, V.A.; Horváth, P. Simple circular dichroism method for selection of the optimal cyclodextrin for drug complexation. J. Incl. Phenom. Macrocycl. Chem. 2019, 95, 223–233. [Google Scholar] [CrossRef]
- Kraszni, M.; Ágh, F.; Horváth, D.; Mirzahosseini, A.; Horváth, P. Effect of Substitution Degree and Homogeneity on Cyclodextrin-Ligand Complex Stability: Comparison of Fenbufen and Fenoprofen Using CD and NMR Spectroscopy. Int. J. Mol. Sci. 2023, 24, 7544. [Google Scholar] [CrossRef]
- Kodaka, M. Application of a general rule to induced circular dichroism of naphthalene derivatives complexed with cyclodextrins. J. Phys. Chem. A 1998, 102, 8101–8103. [Google Scholar] [CrossRef]
- Harata, K.; Uedaira, H. The Circular Dichroism spectra of the β-cyclodextrin complex with naphthalene derivatives. Bull. Chem. Soc. Jpn. 1975, 48, 375–378. [Google Scholar] [CrossRef]
- Kajtár, M.; Horváth-Toró, C.; Kuthi, É.; Szejtli, J. A simple rule for predicting circular dichroism induced in aromatic guests by cyclodextrin hosts in inclusion complexes. In Proceedings of the First International Symposium on Cyclodextrins, Budapest, Hungary, 30 September–2 October 1981; Szejtli, J., Ed.; Springer: Dordrecht, The Netherlands, 1982; pp. 181–193. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Gubica, T. Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. Int. J. Mol. Sci. 2021, 22, 9422. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, L. Current Status of Quantum Chemical Studies of Cyclodextrin Host–Guest Complexes. Molecules 2022, 27, 3874. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, H.; Csillag, A.; Hancu, G.; Székely-Szentmiklósi, B.; Fülöp, I.; Varga, E.; Grama, L.; Orgován, G. Characterisation of inclusion complexes between bifonazole and different cyclodextrins in solid and solution state. Maced. J. Chem. Chem. Eng. 2017, 36, 81–91. [Google Scholar] [CrossRef]
- Bratu, I.; Gavira-Vallejo, J.M.; Hernanz, A.; Bogdan, M.; Bora, G. Inclusion complex of fenbufen with β-cyclodextrin. Biopolymers 2004, 73, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, 11–17 November 2006; Available online: http://dl.acm.org/citation.cfm?id=1188455 (accessed on 30 November 2023).
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Chen, L.Y. Crystal-structures of inclusion complexes of beta-cyclodextrin with (s)-(+)-fenoprofen and (r)-(−)-fenoprofen. J. Am. Chem. Soc. 1988, 110, 4379. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transition Dipole Moment (D) | ||||
|---|---|---|---|---|
| X | Y | Z | Total | |
| Fenoprofen (water) | 0.0917 | −0.5624 | −0.4419 | 0.7211 |
| Fenoprofen–BCyD | −0.0456 | −0.0700 | 0.1518 | 0.1732 |
| Fenbufen (water) | −0.9793 | −0.8206 | −0.4739 | 1.3627 |
| Febufen–BCyD | 0.0105 | −0.1213 | 0.2067 | 0.2398 |
| Nimesulide (water) | 0.5110 | 0.2802 | 0.2288 | 0.6261 |
| Nimesulide–BCyD | −0.5166 | −0.3848 | −1.3888 | 1.5309 |
| Bifonazole (water) | −0.3578 | −0.2423 | −0.0594 | 0.4361 |
| Bifonazole–BCyD biphenyl in ring | −0.0514 | 0.7572 | −0.7827 | 1.0902 |
| Bifonazole–BCyD phenyl in ring | −1.3510 | 1.9622 | −3.2134 | 4.0002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraszni, M.; Balogh, B.; Mándity, I.; Horváth, P. Advantages of Induced Circular Dichroism Spectroscopy for Qualitative and Quantitative Analysis of Solution-Phase Cyclodextrin Host–Guest Complexes. Int. J. Mol. Sci. 2024, 25, 412. https://doi.org/10.3390/ijms25010412
Kraszni M, Balogh B, Mándity I, Horváth P. Advantages of Induced Circular Dichroism Spectroscopy for Qualitative and Quantitative Analysis of Solution-Phase Cyclodextrin Host–Guest Complexes. International Journal of Molecular Sciences. 2024; 25(1):412. https://doi.org/10.3390/ijms25010412
Chicago/Turabian StyleKraszni, Márta, Balázs Balogh, István Mándity, and Péter Horváth. 2024. "Advantages of Induced Circular Dichroism Spectroscopy for Qualitative and Quantitative Analysis of Solution-Phase Cyclodextrin Host–Guest Complexes" International Journal of Molecular Sciences 25, no. 1: 412. https://doi.org/10.3390/ijms25010412