
NADPH Dynamics: Linking Insulin Resistance and β-Cells Ferroptosis in Diabetes Mellitus
Abstract
:1. Introduction
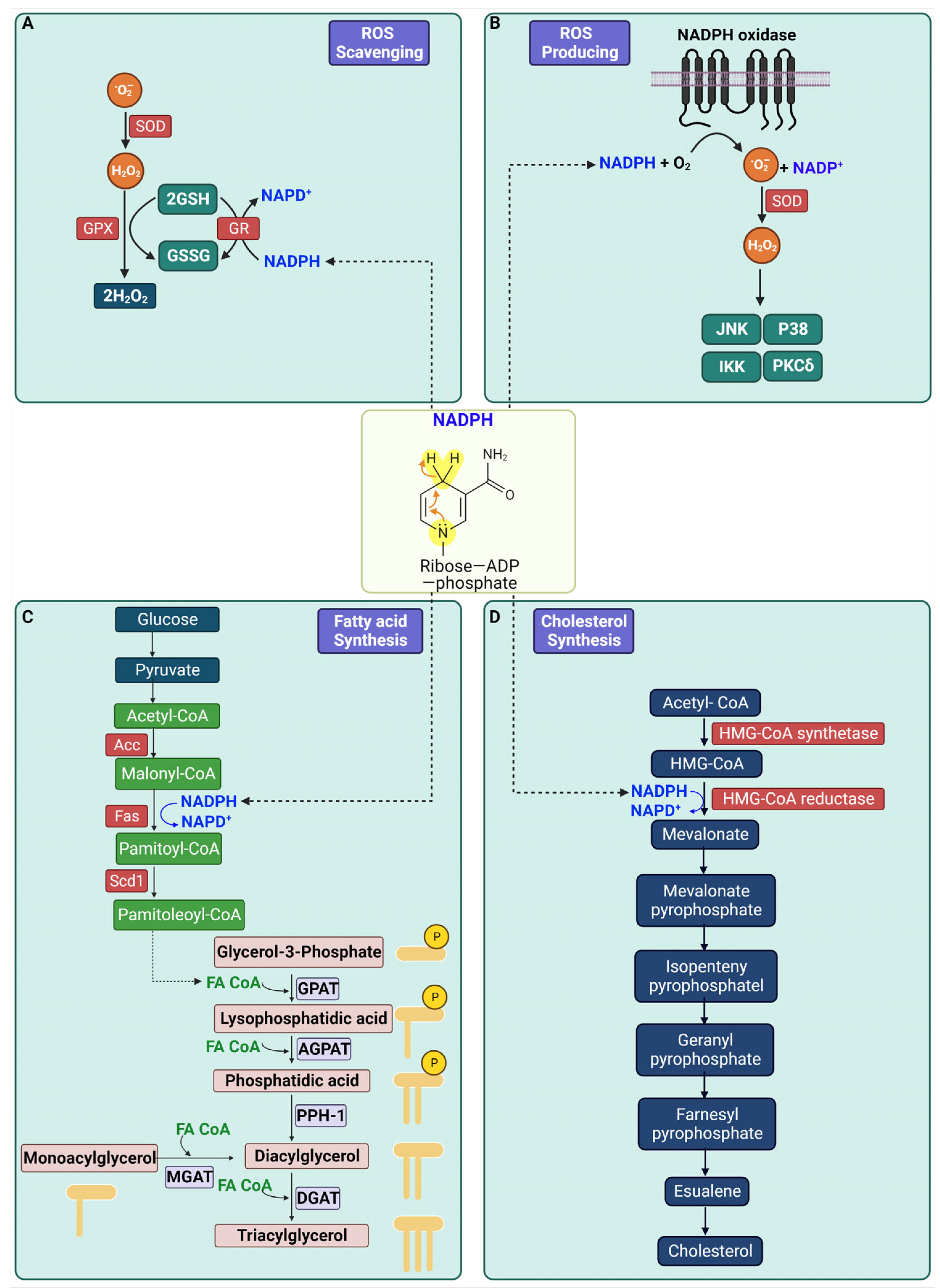
2. Biological Role of NADPH
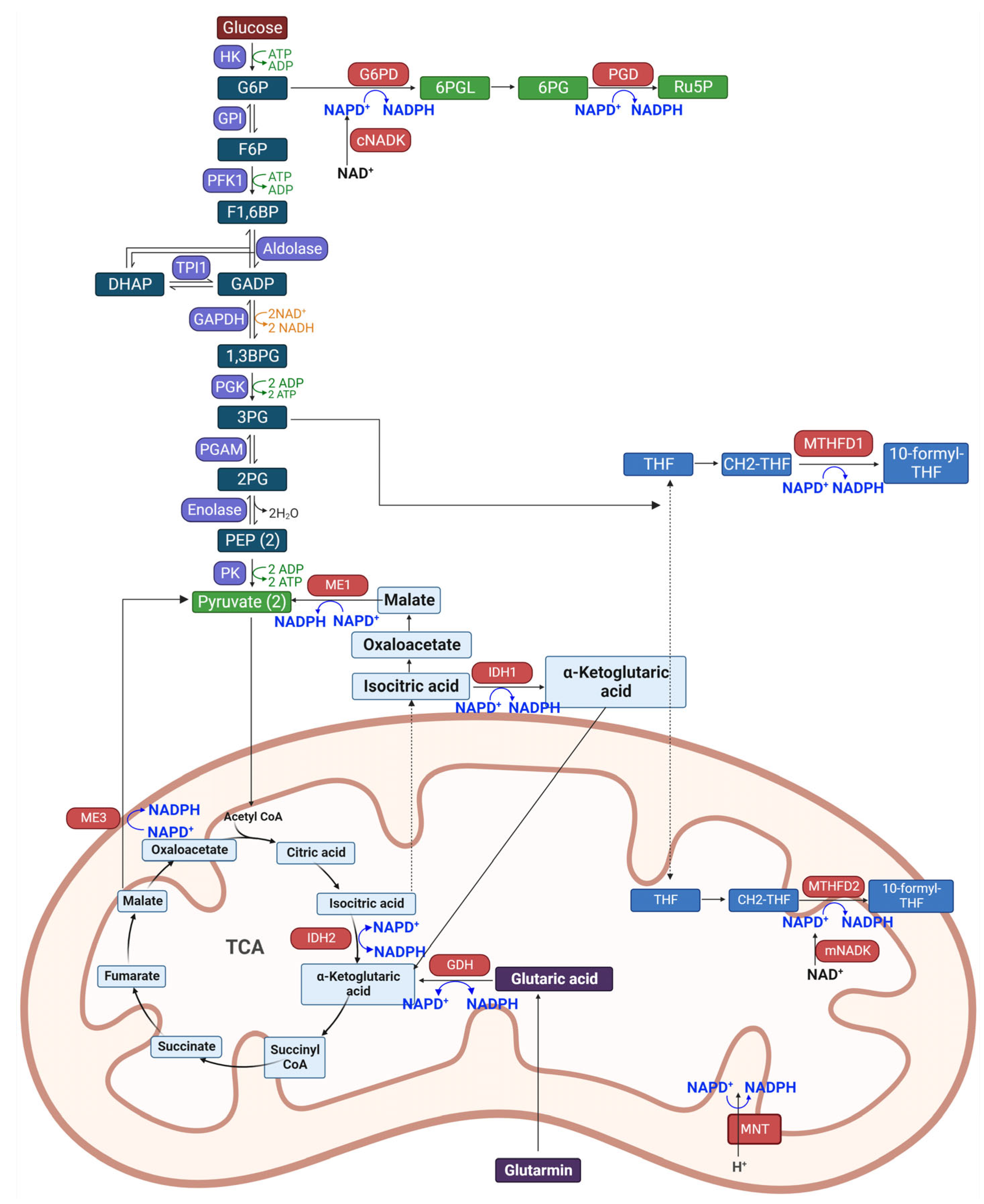
3. Biosynthetic Pathway of NADPH
4. Changes in Expression of NADPH Producing Enzymes in Diabetes Mellitus
5. Role of NADPH-Producing Enzymes in Diabetes
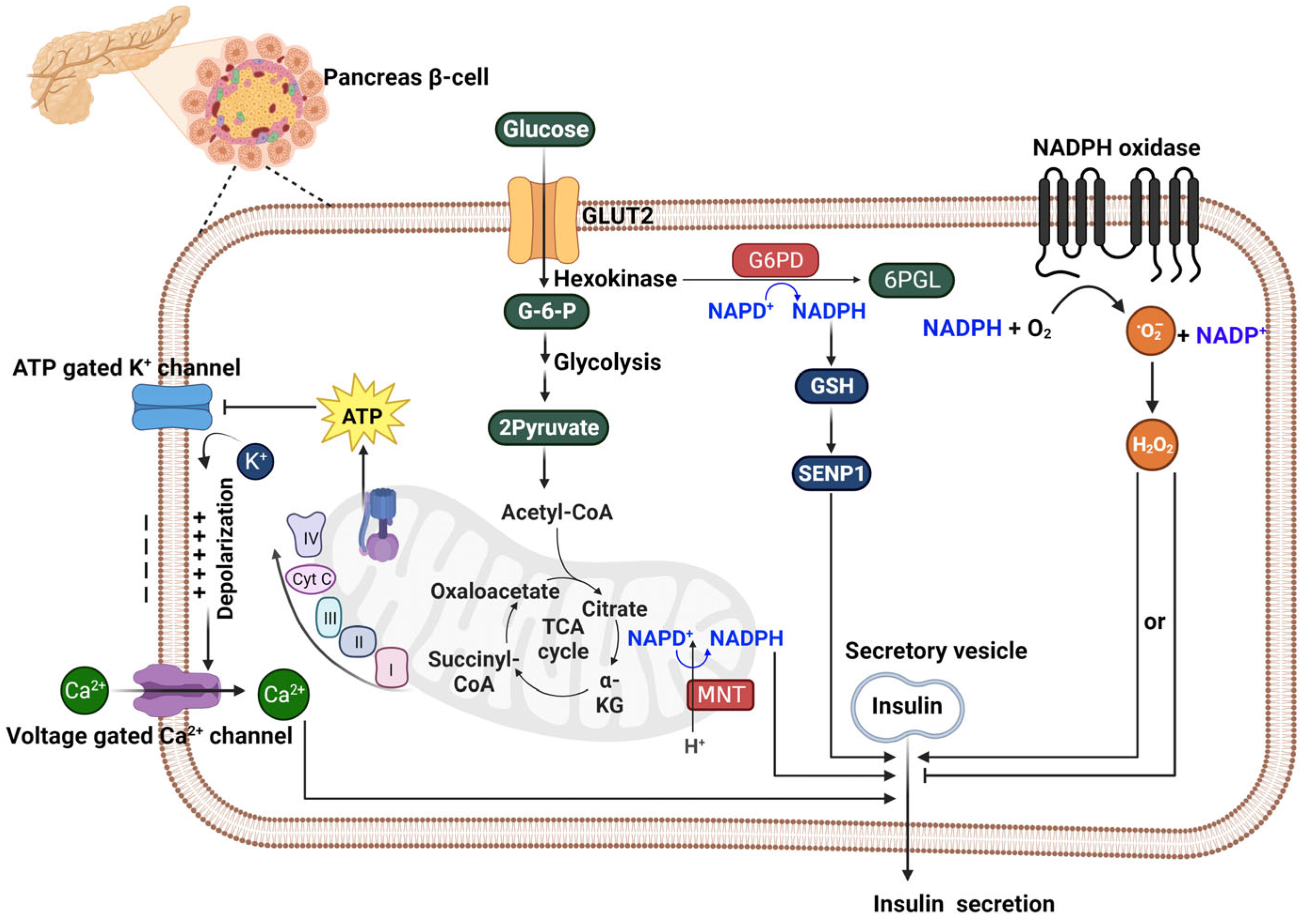
6. The Role of NADPH in Affecting Insulin Secretion
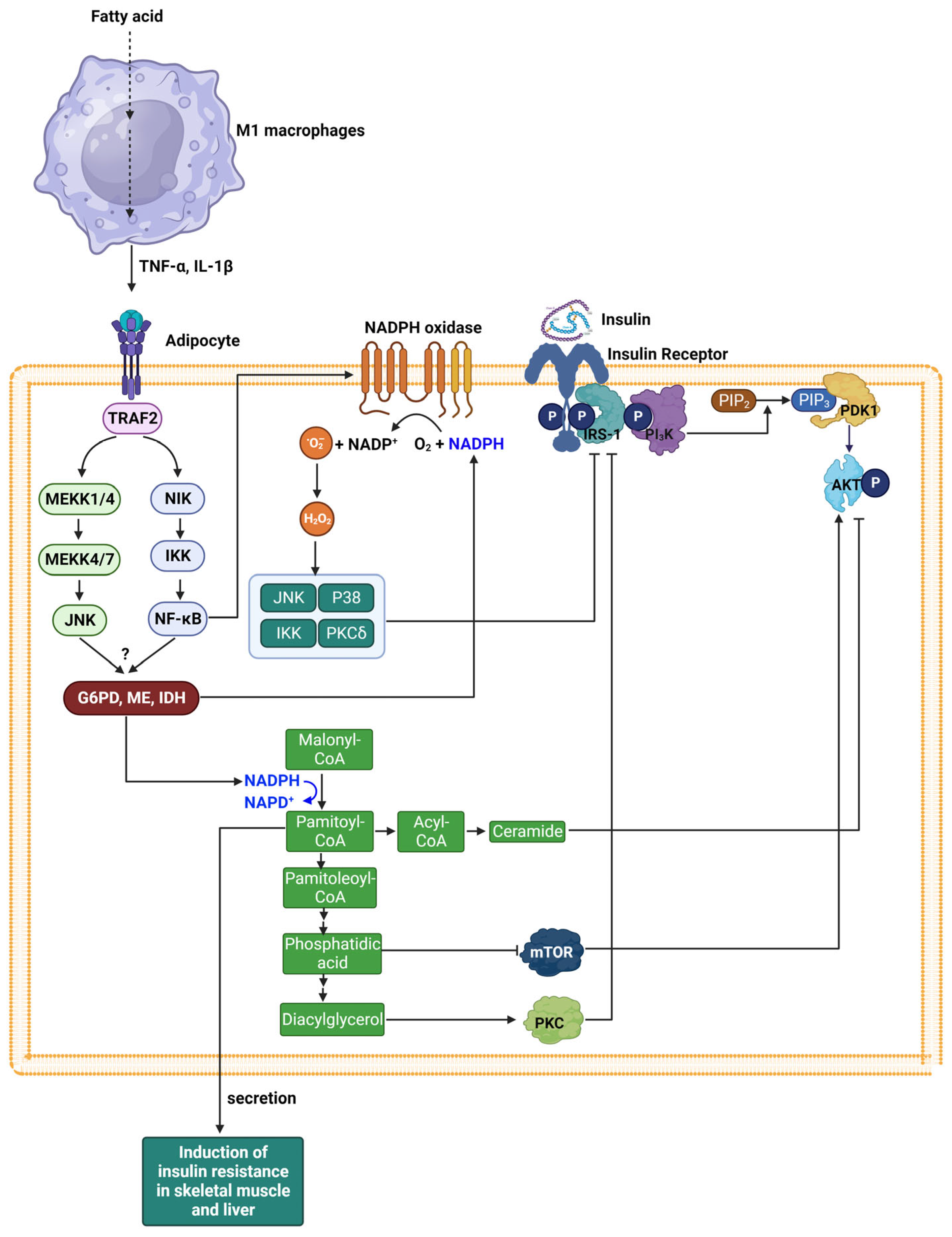
7. Insulin Resistance and NADPH
8. Ferroptosis and NADPH in T2DM
9. The Role of NOXs in Insulin Secretion, Insulin Resistance, and Ferroptosis
10. Conclusions
Funding
Conflicts of Interest
References
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.P.; Duvuuri, V. Diabetes mellitus. Clin. Podiatr. Med. Surg. 2002, 19, 79–107. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B.; Arneth, R.; Shams, M. Metabolomics of Type 1 and Type 2 Diabetes. Int. J. Mol. Sci. 2019, 20, 2467. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Yang, Y.; Zhang, L.; Sun, Z.; Jia, G.; Parrish, A.R.; Sowers, J.R. Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism 2021, 119, 154766. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. S2), S135–S148. [Google Scholar] [CrossRef]
- Nascè, A.; Gariani, K.; Jornayvaz, F.R.; Szanto, I. NADPH Oxidases Connecting Fatty Liver Disease, Insulin Resistance and Type 2 Diabetes: Current Knowledge and Therapeutic Outlook. Antioxidants 2022, 11, 1131. [Google Scholar] [CrossRef]
- Gray, J.P.; Alavian, K.N.; Jonas, E.A.; Heart, E.A. NAD kinase regulates the size of the NADPH pool and insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E191–E199. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Onyango, A.N. Cellular Stresses and Stress Responses in the Pathogenesis of Insulin Resistance. Oxid. Med. Cell Longev. 2018, 2018, 4321714. [Google Scholar] [CrossRef] [PubMed]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824. [Google Scholar]
- García, J.G.; Ansorena, E.; Izal, I.; Zalba, G.; de Miguel, C.; Milagro, F.I. Structure, regulation, and physiological functions of NADPH oxidase 5 (NOX5). J. Physiol. Biochem. 2023, 79, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Grune, T.; Speckmann, B. The two faces of reactive oxygen species (ROS) in adipocyte function and dysfunction. Biol. Chem. 2016, 397, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef]
- Vilas-Boas, E.A.; Almeida, D.C.; Roma, L.P.; Ortis, F.; Carpinelli, A.R. Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells 2021, 10, 3328. [Google Scholar] [CrossRef]
- Gałecka, E.; Jacewicz, R.; Mrowicka, M.; Florkowski, A.; Gałecki, P. Antioxidative enzymes—Structure, properties, functions. Pol. Merkur. Lekarski 2008, 25, 266–268. [Google Scholar]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Pearce, J. Fatty acid synthesis in liver and adipose tissue. Proc. Nutr. Soc. 1983, 42, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [PubMed]
- Cerqueira, N.M.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef] [PubMed]
- Pramono, A.A.; Rather, G.M.; Herman, H.; Lestari, K.; Bertino, J.R. NAD- and NADPH-Contributing Enzymes as Therapeutic Targets in Cancer: An Overview. Biomolecules 2020, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Kawai, S.; Murata, K. Identification and characterization of a human mitochondrial NAD kinase. Nat. Commun. 2012, 3, 1248. [Google Scholar] [CrossRef]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Du, W.; Jiang, P.; Mancuso, A.; Stonestrom, A.; Brewer, M.D.; Minn, A.J.; Mak, T.W.; Wu, M.; Yang, X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat. Cell Biol. 2013, 15, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Moran, D.M.; Trusk, P.B.; Pry, K.; Paz, K.; Sidransky, D.; Bacus, S.S. KRAS mutation status is associated with enhanced dependency on folate metabolism pathways in non-small cell lung cancer cells. Mol. Cancer Ther. 2014, 13, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Zsippai, A.; Szabó, D.R.; Tömböl, Z.; Szabó, P.M.; Eder, K.; Pállinger, E.; Gaillard, R.C.; Patócs, A.; Tóth, S.; Falus, A.; et al. Effects of mitotane on gene expression in the adrenocortical cell line NCI-H295R: A microarray study. Pharmacogenomics 2012, 13, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Ackerstaff, E.; Gimi, B.; Artemov, D.; Bhujwalla, Z.M. Anti-inflammatory agent indomethacin reduces invasion and alters metabolism in a human breast cancer cell line. Neoplasia 2007, 9, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Piva, R. Wild-Type IDH Enzymes as Actionable Targets for Cancer Therapy. Cancers 2019, 11, 563. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Nakayama, Y.; Fukusaki, E.; Irino, Y. Glutamate production from ammonia via glutamate dehydrogenase 2 activity supports cancer cell proliferation under glutamine depletion. Biochem. Biophys. Res. Commun. 2018, 495, 761–767. [Google Scholar] [CrossRef]
- Fernandes, L.M.; Al-Dwairi, A.; Simmen, R.C.M.; Marji, M.; Brown, D.M.; Jewell, S.W.; Simmen, F.A. Malic Enzyme 1 (ME1) is pro-oncogenic in Apc(Min/+) mice. Sci. Rep. 2018, 8, 14268. [Google Scholar] [CrossRef]
- Murphy, M.P. Redox Modulation by Reversal of the Mitochondrial Nicotinamide Nucleotide Transhydrogenase. Cell Metab. 2015, 22, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.P.; Kang, Y.P.; Falzone, A.; Boyle, T.A.; DeNicola, G.M. Nicotinamide nucleotide transhydrogenase regulates mitochondrial metabolism in NSCLC through maintenance of Fe-S protein function. J. Exp. Med. 2020, 217, e20191689. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Flores, M.; Ibáñez-Hernández, M.A.; Galván, R.E.; Gutiérrez, M.; Durán-Reyes, G.; Medina-Navarro, R.; Pascoe-Lira, D.; Ortega-Camarillo, C.; Vilar-Rojas, C.; Cruz, M.; et al. Glucose-6-phosphate dehydrogenase activity and NADPH/NADP+ ratio in liver and pancreas are dependent on the severity of hyperglycemia in rat. Life Sci. 2006, 78, 2601–2607. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Ha, S.O.; Koh, H.J.; Kim, K.; Jeon, S.M.; Choi, M.S.; Kwon, O.S.; Huh, T.L. Upregulation of cytosolic NADP+-dependent isocitrate dehydrogenase by hyperglycemia protects renal cells against oxidative stress. Mol. Cells 2010, 29, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Karunakaran, U.; Moon, J.S.; Won, K.C. NADPH Oxidase (NOX) Targeting in Diabetes: A Special Emphasis on Pancreatic β-Cell Dysfunction. Cells 2021, 10, 1573. [Google Scholar] [CrossRef] [PubMed]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.A.; Jung, S.H.; Jeon, H.Y.; Lee, Y.J.; Lee, J.Y.; Han, E.T.; Park, W.S.; Hong, S.H.; Kim, Y.M.; Ha, K.S. Activity-expression profiling of glucose-6-phosphate dehydrogenase in tissues of normal and diabetic mice. Biochem. Biophys. Res. Commun. 2020, 524, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Fomina, M.P. Effect of starvation and diabetes on the activity of glucose-6-phosphate and 6-phosphogluconate dehydrogenases and on the free fatty acid content of rat kidney cortex and medulla. Vopr. Med. Khim 1978, 24, 377–382. [Google Scholar]
- Ulusu, N.N.; Ozbey, G.; Tandogan, B.; Gunes, A.; Durakoglugil, D.B.; Karasu, C.; Uluoglu, C.; Zengil, H. Circadian variations in the activities of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in the liver of control and streptozotocin-induced diabetic rats. Chronobiol. Int. 2005, 22, 667–677. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Q. Three genes expressed in relation to lipid metabolism considered as potential biomarkers for the diagnosis and treatment of diabetic peripheral neuropathy. Sci. Rep. 2023, 13, 8679. [Google Scholar] [CrossRef]
- Li, H.; Shi, X.; Jiang, H.; Kang, J.; Yu, M.; Li, Q.; Yu, K.; Chen, Z.; Pan, H.; Chen, W. CMap analysis identifies Atractyloside as a potential drug candidate for type 2 diabetes based on integration of metabolomics and transcriptomics. J. Cell Mol. Med. 2020, 24, 7417–7426. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xiong, Q.; He, G.; Tang, J.; Sun, L.; Cheng, S.; Ke, M.; Chen, S.; Hu, Y.; Feng, J.; et al. Hepatic IDH2 regulates glycolysis and gluconeogenesis. Metabolism 2023, 143, 155559. [Google Scholar] [CrossRef] [PubMed]
- Vohra, M.; Adhikari, P.; Souza, S.C.; Nagri, S.K.; Umakanth, S.; Satyamoorthy, K.; Rai, P.S. CpG-SNP site methylation regulates allele-specific expression of MTHFD1 gene in type 2 diabetes. Lab. Investig. 2020, 100, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- King, M.J.; Pugazhenthi, S.; Khandelwal, R.L.; Sharma, R.K. Elevated N-myristoyl transferase activity is reversed by sodium orthovanadate in streptozotocin-induced diabetic rat. Biochim. Biophys. Acta 1993, 1165, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, A.; Guo, H.; Jiang, X.; Tao, J.; Wu, W.; Liu, H. G6PD Deficiency Is Crucial for Insulin Signaling Activation in Skeletal Muscle. Int. J. Mol. Sci. 2022, 23, 7425. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, Y.; Niu, Y.; Wang, C.; Wang, M.; Li, Y.; Sun, C. Activated glucose-6-phosphate dehydrogenase is associated with insulin resistance by upregulating pentose and pentosidine in diet-induced obesity of rats. Horm. Metab. Res. 2012, 44, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Rho, H.K.; Kim, K.H.; Choe, S.S.; Lee, Y.S.; Kim, J.B. Overexpression of glucose-6-phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol. Cell Biol. 2005, 25, 5146–5157. [Google Scholar] [CrossRef] [PubMed]
- Monte Alegre, S.; Saad, S.T.; Delatre, E.; Saad, M.J. Insulin secretion in patients deficient in glucose-6-phosphate dehydrogenase. Horm. Metab. Res. 1991, 23, 171–173. [Google Scholar] [CrossRef]
- Lee, J.W.; Choi, A.H.; Ham, M.; Kim, J.W.; Choe, S.S.; Park, J.; Lee, G.Y.; Yoon, K.H.; Kim, J.B. G6PD up-regulation promotes pancreatic beta-cell dysfunction. Endocrinology 2011, 152, 793–803. [Google Scholar] [CrossRef]
- Spégel, P.; Sharoyko, V.V.; Goehring, I.; Danielsson, A.P.; Malmgren, S.; Nagorny, C.L.; Andersson, L.E.; Koeck, T.; Sharp, G.W.; Straub, S.G.; et al. Time-resolved metabolomics analysis of β-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem. J. 2013, 450, 595–605. [Google Scholar] [CrossRef]
- Al-Dwairi, A.; Brown, A.R.; Pabona, J.M.; Van, T.H.; Hamdan, H.; Mercado, C.P.; Quick, C.M.; Wight, P.A.; Simmen, R.C.; Simmen, F.A. Enhanced gastrointestinal expression of cytosolic malic enzyme (ME1) induces intestinal and liver lipogenic gene expression and intestinal cell proliferation in mice. PLoS ONE 2014, 9, e113058. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.M.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; MacDonald, M.J. Mitochondrial malic enzyme 3 is important for insulin secretion in pancreatic β-cells. Mol. Endocrinol. 2015, 29, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Joly, E.; Pepin, E.; Barbeau, A.; Hentsch, L.; Pineda, M.; Madiraju, S.R.; Brunengraber, H.; Prentki, M. A role for cytosolic isocitrate dehydrogenase as a negative regulator of glucose signaling for insulin secretion in pancreatic ß-cells. PLoS ONE 2013, 8, e77097. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, S.H.; Park, K.M.; Lee, J.H.; Park, J.W. Increased obesity resistance and insulin sensitivity in mice lacking the isocitrate dehydrogenase 2 gene. Free Radic. Biol. Med. 2016, 99, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Fu, Z.; Yang, Z.; Song, Z.; Shamsa, E.H.; Yumnamcha, T.; Sun, S.; Liu, W.; Ibrahim, A.S.; Qi, N.R.; et al. The mitochondrial NAD kinase functions as a major metabolic regulator upon increased energy demand. Mol. Metab. 2022, 64, 101562. [Google Scholar] [CrossRef] [PubMed]
- Vetterli, L.; Carobbio, S.; Frigerio, F.; Karaca, M.; Maechler, P. The Amplifying Pathway of the β-Cell Contributes to Diet-induced Obesity. J. Biol. Chem. 2016, 291, 13063–13075. [Google Scholar] [CrossRef] [PubMed]
- Petraki, Z.; Droubogiannis, S.; Mylonaki, K.; Chlouverakis, G.; Plaitakis, A.; Spanaki, C. Transgenic expression of the positive selected human GLUD2 gene improves in vivo glucose homeostasis by regulating basic insulin secretion. Metabolism 2019, 100, 153958. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Li, W.; Huang, E.; Gao, S. Type 1 Diabetes Mellitus and Cognitive Impairments: A Systematic Review. J. Alzheimers Dis. 2017, 57, 29–36. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Merrins, M.J.; Corkey, B.E.; Kibbey, R.G.; Prentki, M. Metabolic cycles and signals for insulin secretion. Cell Metab. 2022, 34, 947–968. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.; Satin, L.S. Beta-Cell Ion Channels and Their Role in Regulating Insulin Secretion. Compr. Physiol. 2021, 11, 1–21. [Google Scholar] [PubMed]
- Kalwat, M.A.; Cobb, M.H. Mechanisms of the amplifying pathway of insulin secretion in the β cell. Pharmacol. Ther. 2017, 179, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Ferdaoussi, M.; Dai, X.; Jensen, M.V.; Wang, R.; Peterson, B.S.; Huang, C.; Ilkayeva, O.; Smith, N.; Miller, N.; Hajmrle, C.; et al. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Investig. 2015, 125, 3847–3860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liew, C.W.; Handy, D.E.; Zhang, Y.; Leopold, J.A.; Hu, J.; Guo, L.; Kulkarni, R.N.; Loscalzo, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J. 2010, 24, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; Jabůrek, M.; Holendová, B.; Tauber, J.; Pavluch, V.; Berková, Z.; Cahová, M.; Schröder, K.; Brandes, R.P.; Siemen, D.; et al. Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes 2020, 69, 1341–1354. [Google Scholar] [CrossRef]
- Li, N.; Li, B.; Brun, T.; Deffert-Delbouille, C.; Mahiout, Z.; Daali, Y.; Ma, X.J.; Krause, K.H.; Maechler, P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012, 61, 2842–2850. [Google Scholar] [CrossRef]
- Santos, L.R.B.; Muller, C.; de Souza, A.H.; Takahashi, H.K.; Spégel, P.; Sweet, I.R.; Chae, H.; Mulder, H.; Jonas, J.C. NNT reverse mode of operation mediates glucose control of mitochondrial NADPH and glutathione redox state in mouse pancreatic β-cells. Mol. Metab. 2017, 6, 535–547. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Pessin, J.E.; Saltiel, A.R. Signaling pathways in insulin action: Molecular targets of insulin resistance. J. Clin. Investig. 2000, 106, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.K.; Jun, W.; Lee, J. Mechanism of ER Stress and Inflammation for Hepatic Insulin Resistance in Obesity. Ann. Nutr. Metab. 2015, 67, 218–227. [Google Scholar] [CrossRef]
- Han, C.Y.; Umemoto, T.; Omer, M.; Den Hartigh, L.J.; Chiba, T.; LeBoeuf, R.; Buller, C.L.; Sweet, I.R.; Pennathur, S.; Abel, E.D.; et al. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 2012, 287, 10379–10393. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.E.; Rey, F.E.; Carretero, O.A.; Pagano, P.J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2234–H2240. [Google Scholar] [CrossRef]
- Dinauer, M.C.; Orkin, S.H.; Brown, R.; Jesaitis, A.J.; Parkos, C.A. The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome b complex. Nature 1987, 327, 717–720. [Google Scholar] [CrossRef]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Shulman, G.I. Unraveling the cellular mechanism of insulin resistance in humans: New insights from magnetic resonance spectroscopy. Physiology 2004, 19, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Klett, E.L.; Coleman, R.A. Lipid signals and insulin resistance. Clin. Lipidol. 2013, 8, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef]
- Li, Y.; Ding, L.; Hassan, W.; Abdelkader, D.; Shang, J. Adipokines and hepatic insulin resistance. J. Diabetes Res. 2013, 2013, 170532. [Google Scholar] [CrossRef] [PubMed]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal 2017, 26, 501–518. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Sosunov, S.A.; Shakerley, N.L.; Ten, V.S.; Ratner, A.J. Hyperglycemic Conditions Prime Cells for RIP1-dependent Necroptosis. J. Biol. Chem. 2016, 291, 13753–13761. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis. Cell Chem. Biol. 2019, 26, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Bauldry, S.A.; Nasrallah, V.N.; Bass, D.A. Activation of NADPH oxidase in human neutrophils permeabilized with Staphylococcus aureus alpha-toxin. A lower Km when the enzyme is activated in situ. J. Biol. Chem. 1992, 267, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Ulusu, N.N.; Tandoğan, B. Purification and kinetic properties of glutathione reductase from bovine liver. Mol. Cell Biochem. 2007, 303, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Anwer, T.; Sharma, M.; Pillai, K.K.; Khan, G. Protective effect of Withania somnifera against oxidative stress and pancreatic beta-cell damage in type 2 diabetic rats. Acta Pol. Pharm. 2012, 69, 1095–1101. [Google Scholar]
- Imoto, H.; Sasaki, N.; Iwase, M.; Nakamura, U.; Oku, M.; Sonoki, K.; Uchizono, Y.; Iida, M. Impaired insulin secretion by diphenyleneiodium associated with perturbation of cytosolic Ca2+ dynamics in pancreatic beta-cells. Endocrinology 2008, 149, 5391–5400. [Google Scholar] [CrossRef]
- Morgan, D.; Rebelato, E.; Abdulkader, F.; Graciano, M.F.; Oliveira-Emilio, H.R.; Hirata, A.E.; Rocha, M.S.; Bordin, S.; Curi, R.; Carpinelli, A.R. Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic beta-cells. Endocrinology 2009, 150, 2197–2201. [Google Scholar] [CrossRef]
- Bouzakri, K.; Veyrat-Durebex, C.; Holterman, C.; Arous, C.; Barbieux, C.; Bosco, D.; Altirriba, J.; Alibashe, M.; Tournier, B.B.; Gunton, J.E.; et al. Beta-Cell-Specific Expression of Nicotinamide Adenine Dinucleotide Phosphate Oxidase 5 Aggravates High-Fat Diet-Induced Impairment of Islet Insulin Secretion in Mice. Antioxid. Redox Signal 2020, 32, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Souto Padron de Figueiredo, A.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef] [PubMed]
- Den Hartigh, L.J.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.S.; Floyd, B.C.; Kozicky, M.; George, S.; Ungvari, Z.I.; Neito, V.; Wolin, M.S.; Gupte, S.A. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic. Biol. Med. 2009, 47, 219–228. [Google Scholar] [CrossRef]
- Park, J.H.; Yoon, S.G.; Ghee, J.Y.; Yoo, J.A.; Cha, J.J.; Kang, Y.S.; Han, S.Y.; Seol, Y.J.; Han, J.Y.; Cha, D.R. Pan-Nox inhibitor treatment improves renal function in aging murine diabetic kidneys. Kidney Res. Clin. Pract. 2023. [Google Scholar] [CrossRef]
- Yao, W.; Liao, H.; Pang, M.; Pan, L.; Guan, Y.; Huang, X.; Hei, Z.; Luo, C.; Ge, M. Inhibition of the NADPH Oxidase Pathway Reduces Ferroptosis during Septic Renal Injury in Diabetic Mice. Oxid. Med. Cell Longev. 2022, 2022, 1193734. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell Mol. Med. 2020, 24, 6670–6679. [Google Scholar] [CrossRef]
- Weaver, J.R.; Holman, T.R.; Imai, Y.; Jadhav, A.; Kenyon, V.; Maloney, D.J.; Nadler, J.L.; Rai, G.; Simeonov, A.; Taylor-Fishwick, D.A. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell Endocrinol. 2012, 358, 88–95. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | mRNA | Protein | Activity | Model | Ref. |
|---|---|---|---|---|---|
| G6PD | NT | Elevated G6PD expression in kidney (2.0-fold), lung (1.2-fold), spleen (1.2-fold), and thyroid (3.2-fold) | Elevated G6PD activities in kidney (10.1-fold), liver (1.5-fold), spleen (2.1-fold), and thyroid (3.3-fold) | Streptozotocin-induced diabetic mice | [47,48] |
| NT | NT | Elevated activity in kidney (2.0-fold) | |||
| PGD | NT | NT | Elevated enzyme activity | Streptozotocin-induced diabetic mice | [49] |
| ME1 | Elevated expression | NT | NT | Diabetic peripheral neuropathy patients | [50,51] |
| NT | Elevated expression | NT | db/db mice | ||
| ME2 | NT | Elevated expression | NT | db/db mice | [51] |
| IDH1 | NT | Elevated expression | Elevated activity | Diabetic rats and mice | [44,52] |
| IDH2 | NT | Elevated expression | NT | High-fat diet mice | [44,52] |
| MTHFD1 | Elevated expression (2.9-fold) | NT | NT | T2D patients | [53] |
| MNT | NT | NT | Elevated activity (2.0-fold) | Streptozotocin-induced diabetic mice | [54] |
| Enzymes | Model | Action | Result | Ref. |
|---|---|---|---|---|
| G6PD | C2C12 mouse myoblast cell line | Over-Expression | Insulin resistance ↑ | [55] |
| Wistar rats | Over-Expression by high fat diet | Insulin resistance ↑ | [56] | |
| Adipocytes | Over-Expression | Insulin resistance ↑ | [57] | |
| Patients with G6PD deficiency | Over-Expression | Insulin secretion ↓ | [58] | |
| Pancreatic β-cells. | Over-Expression | Insulin secretion ↓ | [59] | |
| 6PGD | β cells | Glucose stimulation | Insulin secretion ↓ | [60] |
| ME1 | Transgenic(Tg) mice | Over-Expression | Insulin resistance ↑ | [61] |
| ME3 | CHS cell line | Knock down (si-RNA) | Insulin release ↓ | [62] |
| IDH1 | INS-1 832/13 cell line | Knockdown | Insulin release ↓ | [63] |
| IDH2 | Mice | Knockout (IDH2−/−) | Insulin resistance ↓ | [64] |
| (Insulin sensitivity ↑) | ||||
| INS-1 832/13 cell line | IDH2 knockdown | Insulin release ↓ | [52] | |
| NAD kinase | Mice | Knockout | Insulin resistance ↑ | [65] |
| GDH1 | Mice | Knockout | Diet-induced Obesity ↓ | [66] |
| GDH2 | Mice | Knockin | Fasting serum insulin levels ↑ | [67] |
| Model | Method | Result | Ref. |
|---|---|---|---|
| Rat Pancreatic Islets, RINm5F cells | Treatment with DPI (NOX inhibitor) | Suppressed insulin secretion in response to high glucose | [109] |
| Rat Pancreatic Islets | Treatment with DPI; Antisense Oligonucleotide for p47(PHOX) | Decreased ROS production, inhibited glucose-stimulated insulin secretion, reduced glucose oxidation | [110] |
| Pancreatic Islets, INS-1E Cells | Knockout (NOX4βKO), Silencing (NOX4) | Suppressed insulin secretion in response to high glucose | [78] |
| Mouse and Human Pancreatic Islets | Nox2 Isoform-Specific Knockout; In Vitro Knockdown of Nox2 | Potentiation of insulin release in Nox2-deficient islets compared with controls | [79] |
| Human Islets | NOX5 knockdown | Suppressed insulin secretion in response to high glucose | [111] |
| Nox2-null Mice; C2C12 Myotubes | Nox2 Knockout; shRNA Down-regulation of Nox2 in Cells | Insulin resistance induced by high-fat diet was reduced in Nox2-null mice compared to wild-type. Nox2 knockdown in C2C12 cells prevented insulin resistance induced by high glucose or palmitate. | [112] |
| C57BL/6 Mice with Specific Adipocyte NOX4 Deletion | NOX4 Knockout in Adipocytes | Delayed insulin resistance | [113] |
| Obese Mice, Human Subjects, Cultured Adipocytes | Treatment with DPI | Improved insulin resistance and reduced oxidative stress | [114,115] |
| Aging Diabetic Mice | Treatment with APX-115 (pan-Nox inhibitor) | Improved insulin resistance and reduced oxidative stress | [116] |
| High-fat diet-fed Mice | Treatment with Vas2870 (NOX inhibitor) | Reduced ferroptosis accumulation and related protein expression (ASCL4, FTH1, and GPX4) | [117] |
| Rats | Nox2-siRNA | Reduced ferroptosis | [118] |
| Human Donor Islets, Mouse Islets, Beta Cell Lines | Treatment with apocynin and DPI (NOX inhibitor) | Reduced ROS production and cell death | [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, D.-O. NADPH Dynamics: Linking Insulin Resistance and β-Cells Ferroptosis in Diabetes Mellitus. Int. J. Mol. Sci. 2024, 25, 342. https://doi.org/10.3390/ijms25010342
Moon D-O. NADPH Dynamics: Linking Insulin Resistance and β-Cells Ferroptosis in Diabetes Mellitus. International Journal of Molecular Sciences. 2024; 25(1):342. https://doi.org/10.3390/ijms25010342
Chicago/Turabian StyleMoon, Dong-Oh. 2024. "NADPH Dynamics: Linking Insulin Resistance and β-Cells Ferroptosis in Diabetes Mellitus" International Journal of Molecular Sciences 25, no. 1: 342. https://doi.org/10.3390/ijms25010342