
Polyploid Genome Assembly Provides Insights into Morphological Development and Ascorbic Acid Accumulation of Sauropus androgynus
 , ,
, ,
Abstract
:1. Introduction
2. Results
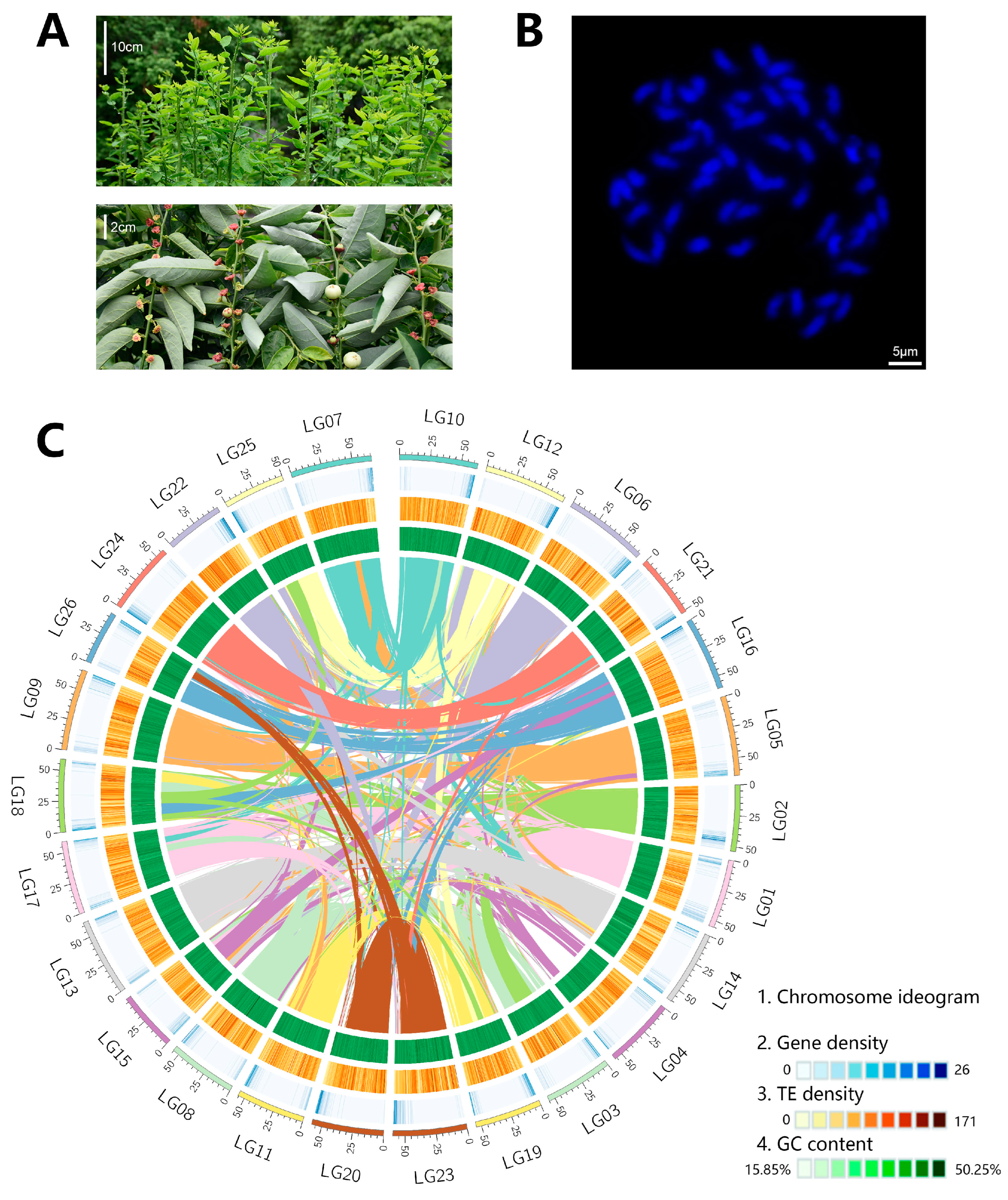
2.1. Potential Polyploid of S. androgynus
2.2. Genome Assembly and Quality Assessment
2.3. LTR Accumulation Promoted the Genome Expansion of S. androgynus
2.4. Gene Prediction and Annotation of S. androgynus
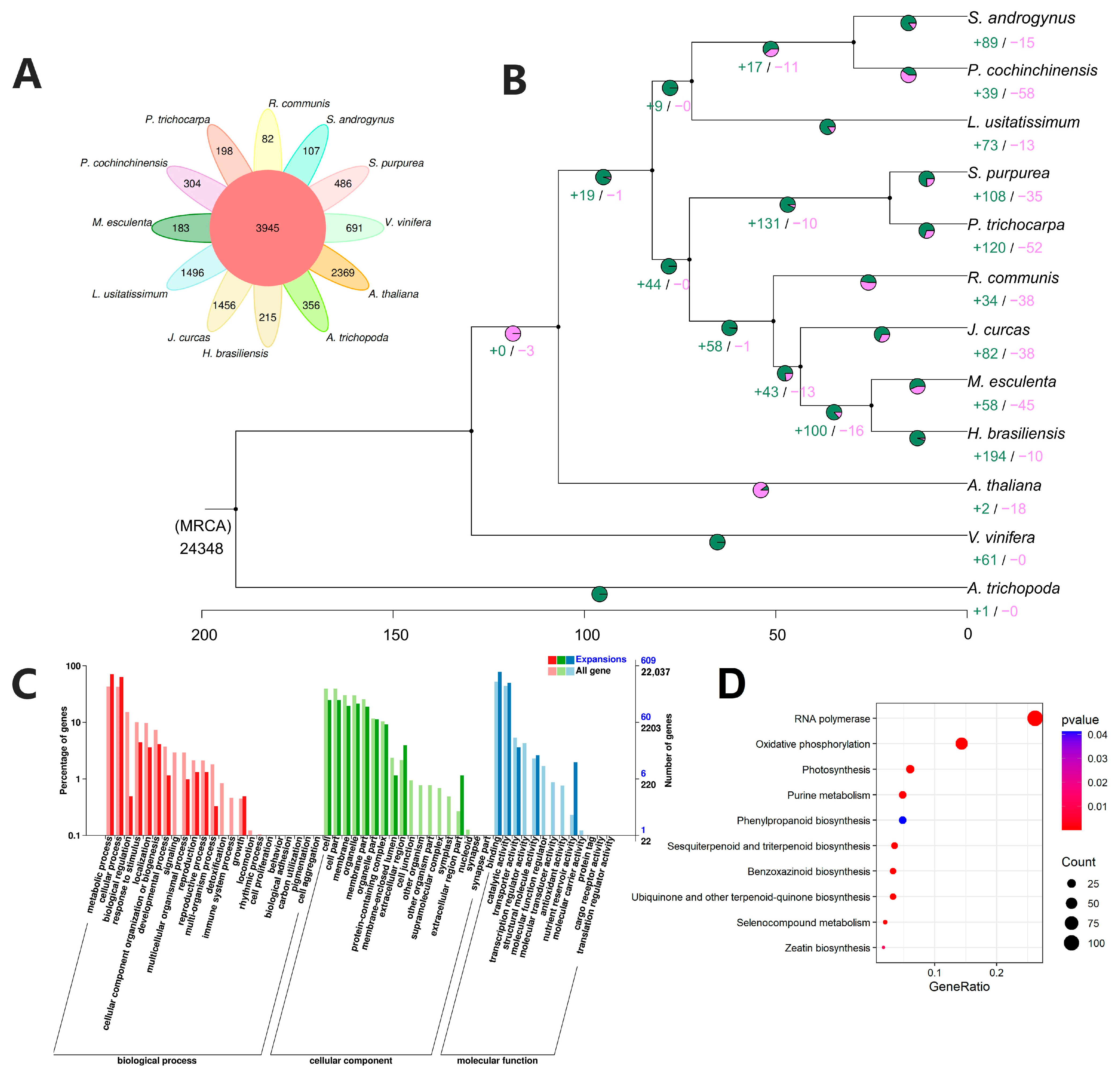
2.5. Evolution History and Comparative Analysis of S. androgynus
2.6. Polyploidization and Synteny Analysis of S. androgynus
2.7. Expansion of Genes Related to Morphological Development
2.8. Transcriptional Regulation of Ascorbic Acid Accumulation in S. androgynus
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sequencing
4.2. Genome Features Estimation
4.3. Genome Assembly by CCS Data
4.4. Hi-C Technology Help Anchor Contigs
4.5. Genome Quality Evaluation
4.6. Annotation of Repetitive Sequences
4.7. Gene Prediction and Annotation
4.8. Gene Family Identification
4.9. Phylogenetic Analysis
4.10. Gene Family Expansion and Contraction Analysis
4.11. WGD Analysis
4.12. Genome Collinearity Analysis
4.13. Transcriptome Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, B.D.; Cheng, J.X.; Zhang, C.F.; Bai, Y.D.; Liu, W.Y.; Li, W.; Koike, K.; Akihisa, T.; Feng, F.; Zhang, J. Sauropus androgynus L. Merr.—A phytochemical, pharmacological and toxicological review. J. Ethnopharmacol. 2020, 257, 112778. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, D.R.; Salim, K.M.; Srivastava, A.; Singh, L.B.; Srivastava, R.C. Estimation of proximate composition, micronutrients and phytochemical compounds in traditional vegetables from Andaman and Nicobar Islands. Int. J. Food Sci. Nutr. 2011, 62, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Neamsuvan, O.; Ruangrit, T. A survey of herbal weeds that are used to treat gastrointestinal disorders from southern Thailand: Krabi and Songkhla provinces. J. Ethnopharmacol. 2017, 209, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Madhu, C.S.; Manukumar, H.M.G.; Basavaraju, P. New-vista in fi nding antioxidant and anti-infl ammatory property of crude protein extract from Sauropus androgynus leaf. Acta Sci. Polon-Technol. 2014, 13, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Nahak, D.G.; Sahu, R. Free Radical Scavenging activity of Multi-vitamin Plant (Sauropus androgynus L. Merr). Researcher 2010, 2, 6–14. [Google Scholar]
- Bunawan, H.; Bunawan, S.N.; Baharum, S.N.; Noor, N.M. Sauropus androgynus (L.) Merr. Induced Bronchiolitis Obliterans: From Botanical Studies to Toxicology. Evid. Based Complement. Alternat. Med. 2015, 2015, 714158. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.J.A. Sauropus androgynus (L.) Merrill-A Potentially Nutritive Functional Leafy-Vegetable. Asian J. Chem. 2013, 25, 9425–9433. [Google Scholar] [CrossRef]
- Padmavathi, P.; Rao, M.P. Nutritive value of Sauropus androgynus leaves. Plant Foods Hum. Nutr. 1990, 40, 107–113. [Google Scholar] [CrossRef]
- Hulshof, P.J.M.; Xu, C.; van de Bovenkamp, P.; Muhilal; West, C.E. Application of a Validated Method for the Determination of Provitamin A Carotenoids in Indonesian Foods of Different Maturity and Origin. J. Agric. Food Chem. 1997, 45, 1174–1179. [Google Scholar] [CrossRef]
- Andarwulan, N.; Kurniasih, D.; Apriady, R.; Rahmat, H.; Roto, A.; Bolling, B. Polyphenols, carotenoids, and ascorbic acid in underutilized medicinal vegetables. J. Funct. Foods 2012, 4, 339–347. [Google Scholar] [CrossRef]
- Miean, K.; Mohamed, S. Flavonoid (Myricetin, Quercetin, Kaempferol, Luteolin, and Apigenin) Content of Edible Tropical Plants. J. Agric. Food Chem. 2001, 49, 3106–3112. [Google Scholar] [CrossRef]
- Kuttinath, S.; Kh, H.; Rammohan, R. Phytochemical screening, antioxidant, antimicrobial, and antibiofilm activity of Sauropus androgynus leaf extracts. Asian J. Pharm. Clin. Res. 2019, 12, 244–250. [Google Scholar] [CrossRef]
- Palombo, E.A.; Semple, S.J. Antibacterial activity of traditional Australian medicinal plants. J. Ethnopharmacol. 2001, 77, 151–157. [Google Scholar] [CrossRef]
- Kathriarachchi, H.; Samuel, R.; Hoffmann, P.; Mlinarec, J.; Wurdack, K.J.; Ralimanana, H.; Stuessy, T.F.; Chase, M.W. Phylogenetics of tribe Phyllantheae (Phyllanthaceae; Euphorbiaceae sensu lato) based on nrITS and plastid matK DNA sequence data. Am. J. Bot. 2006, 93, 637–655. [Google Scholar] [CrossRef]
- Webster, G.L. Synopsis of the Genera and Suprageneric Taxa of Euphorbiaceae. Ann. Mo. Bot. Gard. 1994, 81, 33–144. [Google Scholar] [CrossRef]
- GROUP, T.A.P. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 2003, 141, 399–436. [Google Scholar] [CrossRef]
- Ekasari, W.; Fatmawati, D.; Khoiriah, S.M.; Baqiuddin, W.A.; Nisa, H.Q.; Maharupini, A.A.S.; Wahyuni, T.S.; Oktarina, R.D.; Suhartono, E.; Sahu, R.K. Antimalarial Activity of Extract and Fractions of Sauropus androgynus (L.) Merr. Scientifica 2022, 2022, 3552491. [Google Scholar] [CrossRef]
- Mustarichie, R.; Salsabila, T.; Iskandar, Y. Determination of the Major Component of Water Fraction of Katuk (Sauropus androgynous (L.) Merr.) Leaves by Liquid Chromatography-Mass Spectrometry. J. Pharm. Bioallied Sci. 2019, 11, S611–S618. [Google Scholar] [CrossRef]
- Sattler, M.C.; Carvalho, C.R.; Clarindo, W.R. The polyploidy and its key role in plant breeding. Planta 2016, 243, 281–296. [Google Scholar] [CrossRef]
- Orr-Weaver, T.L. When bigger is better: The role of polyploidy in organogenesis. Trends Genet. 2015, 31, 307–315. [Google Scholar] [CrossRef]
- Chen, R.; Chen, C.; Song, W.; Liang, G.; Li, X.; Chen, L.; Wang, G.; Ma, X.; Wang, W. Chromosome Atlas of Major Eco-Nomic Plants Genome in China Tomus V Chromosome Atlas of Medicinal Plants in China, 1st ed.; Science Press: Beijing, China, 2009; pp. 274–288. [Google Scholar]
- Zhang, W.; Xu, S.; Gu, Y.; Jiao, M.; Mei, Y.; Wang, J. The first high-quality chromosome-level genome assembly of Phyllanthaceae (Phyllanthus cochinchinensis) provides insights into flavonoid biosynthesis. Planta 2022, 256, 109. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef]
- Jiao, Y.; Leebens-Mack, J.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar] [CrossRef]
- Lu, J.-M.; Landrein, S.; Song, X.-Z.; Wu, M.; Xiao, C.-F.; Sun, P.; Jia, H.-Z.; Yue, J.-R.; Xu, Y.-K. Polyploidy leads to phenotypic differences between tetraploid Kaempferia galanga var. latifolia and pentaploid K. galanga var. galanga (Zingiberaceae). Sci. Hortic. 2023, 307, 111527. [Google Scholar] [CrossRef]
- Tavan, M.; Azizi, A.; Sarikhani, H.; Mirjalili, M.H.; Rigano, M.M. Induced polyploidy and broad variation in phytochemical traits and altered gene expression in Salvia multicaulis. Sci. Hortic. 2022, 291, 110592. [Google Scholar] [CrossRef]
- Sugimoto, H.; Tanaka, T.; Muramoto, N.; Kitagawa-Yogo, R.; Mitsukawa, N. Transcription factor NTL9 negatively regulates Arabidopsis vascular cambium development during stem secondary growth. Plant Physiol. 2022, 190, 1731–1746. [Google Scholar] [CrossRef]
- Ragni, L.; Greb, T. Secondary growth as a determinant of plant shape and form. Semin. Cell Dev. Biol. 2018, 79, 58–67. [Google Scholar] [CrossRef]
- Shimizu-Inatsugi, R.; Terada, A.; Hirose, K.; Kudoh, H.; Sese, J.; Shimizu, K.K. Plant adaptive radiation mediated by polyploid plasticity in transcriptomes. Mol. Ecol. 2017, 26, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhu, Y.X. Plant polyploidy and evolution. J. Integr. Plant Biol. 2019, 61, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Rothfels, C.J. Polyploid phylogenetics. New Phytol. 2021, 230, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Chansler, M.T.; Ferguson, C.J.; Fehlberg, S.D.; Prather, L.A. The role of polyploidy in shaping morphological diversity in natural populations of Phlox amabilis. Am. J. Bot. 2016, 103, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Smirnoff, N. Ascorbic acid metabolism and functions: A comparison of plants and mammals. Free Radic Biol. Med. 2018, 122, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Pullar, J.M.; Carr, A.C.; Vissers, M.C.M. The Roles of Vitamin C in Skin Health. Nutrients 2017, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Drouin, G.; Godin, J.R.; Page, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Macknight, R.C.; Laing, W.A.; Bulley, S.M.; Broad, R.C.; Johnson, A.A.; Hellens, R.P. Increasing ascorbate levels in crops to enhance human nutrition and plant abiotic stress tolerance. Curr. Opin. Biotechnol. 2017, 44, 153–160. [Google Scholar] [CrossRef]
- Li, T.; Yang, X.; Yu, Y.; Si, X.; Zhai, X.; Zhang, H.; Dong, W.; Gao, C.; Xu, C. Domestication of wild tomato is accelerated by genome editing. Nat. Biotechnol. 2018, 36, 1160–1163. [Google Scholar] [CrossRef]
- Li, Y.; Liu, G.F.; Ma, L.M.; Liu, T.K.; Zhang, C.W.; Xiao, D.; Zheng, H.K.; Chen, F.; Hou, X.L. A chromosome-level reference genome of non-heading Chinese cabbage [Brassica campestris (syn. Brassica rapa) ssp. chinensis]. Hortic. Res. 2020, 7, 212. [Google Scholar] [CrossRef]
- Feng, C.; Feng, C.; Lin, X.; Liu, S.; Li, Y.; Kang, M. A chromosome-level genome assembly provides insights into ascorbic acid accumulation and fruit softening in guava (Psidium guajava). Plant Biotechnol. J. 2021, 19, 717–730. [Google Scholar] [CrossRef]
- Liu, H.; Wei, L.; Ni, Y.; Chang, L.; Dong, J.; Zhong, C.; Sun, R.; Li, S.; Xiong, R.; Wang, G.; et al. Genome-Wide Analysis of Ascorbic Acid Metabolism Related Genes in Fragaria × ananassa and Its Expression Pattern Analysis in Strawberry Fruits. Front. Plant Sci. 2022, 13, 954505. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Zhao, J.; Cai, Q.L.; Liu, G.C.; Wang, J.R.; Zhao, Z.H.; Liu, P.; Dai, L.; Yan, G.; Wang, W.J.; et al. The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 2014, 5, 5315. [Google Scholar] [CrossRef] [PubMed]
- Deans, R.M.; Brodribb, T.J.; Busch, F.A.; Farquhar, G.D. Plant water-use strategy mediates stomatal effects on the light induction of photosynthesis. New Phytol. 2019, 222, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Braun, H.P. The Oxidative Phosphorylation system of the mitochondria in plants. Mitochondrion 2020, 53, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Cesarino, I.; Eudes, A.; Urbanowicz, B.; Xie, M. Editorial: Phenylpropanoid Systems Biology and Biotechnology. Front. Plant Sci. 2022, 13, 866164. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, W.J.C.; Gruppen, H.; Vincken, J.P. Structure and biosynthesis of benzoxazinoids: Plant defence metabolites with potential as antimicrobial scaffolds. Phytochemistry 2018, 155, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Mei, X.; Wan, S.; Lin, C.; Zhou, C.; Hu, L.; Deng, C.; Zhang, L. Integration of Metabolome and Transcriptome Reveals the Relationship of Benzenoid-Phenylpropanoid Pigment and Aroma in Purple Tea Flowers. Front. Plant Sci. 2021, 12, 762330. [Google Scholar] [CrossRef]
- Wu, Y.; Duan, S.; Zhao, L.; Gao, Z.; Luo, M.; Song, S.; Xu, W.; Zhang, C.; Ma, C.; Wang, S. Aroma characterization based on aromatic series analysis in table grapes. Sci. Rep. 2016, 6, 31116. [Google Scholar] [CrossRef]
- Shang, J.; Tian, J.; Cheng, H.; Yan, Q.; Li, L.; Jamal, A.; Xu, Z.; Xiang, L.; Saski, C.A.; Jin, S.; et al. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 2020, 21, 200. [Google Scholar] [CrossRef]
- Li, Y.; Kong, D.; Fu, Y.; Sussman, M.R.; Wu, H. The effect of developmental and environmental factors on secondary metabolites in medicinal plants. Plant Physiol. Biochem. 2020, 148, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Luo, D.; Zhao, T.; Du, H.; Liu, Z.; Xu, Z.; Guo, L.; Chen, C.; Peng, S.; Li, J.X.; et al. Genome sequencing reveals chromosome fusion and extensive expansion of genes related to secondary metabolism in Artemisia argyi. Plant Biotechnol. J. 2022, 20, 1902–1915. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as Potential Anti-Inflammatory Molecules: A Review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef]
- Jaeger, R.; Cuny, E. Terpenoids with Special Pharmacological Significance: A Review. Nat. Prod. Commun. 2016, 11, 1373–1390. [Google Scholar] [CrossRef]
- Lin, M.; Jian, J.B.; Zhou, Z.Q.; Chen, C.H.; Wang, W.; Xiong, H.; Mei, Z.N. Chromosome-level genome of Entada phaseoloides provides insights into genome evolution and biosynthesis of triterpenoid saponins. Mol. Ecol. Resour. 2022, 22, 3049–3067. [Google Scholar] [CrossRef]
- Abu Almakarem, A.S.; Heilman, K.L.; Conger, H.L.; Shtarkman, Y.M.; Rogers, S.O. Extraction of DNA from plant and fungus tissues in situ. BMC Res. Notes 2012, 5, 266. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Huang, Y.; Ding, W.; Zhang, M.; Han, J.; Jing, Y.; Yao, W.; Hasterok, R.; Wang, Z.; Wang, K. The formation and evolution of centromeric satellite repeats in Saccharum species. Plant J. 2021, 106, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Neumann, P.; Novak, P.; Hostakova, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Clements, J.; Eddy, S.R.; Hubley, R.; Jones, T.A.; Jurka, J.; Smit, A.F.; Finn, R.D. Dfam: A database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013, 41, D70–D82. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 5, 4–10. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol. Biol. 2019, 1962, 161–177. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2008, 36, D281–D288. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology, C. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- She, R.; Chu, J.S.; Wang, K.; Pei, J.; Chen, N. GenBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 2009, 19, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Puttick, M.N. MCMCtreeR: Functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 2019, 35, 5321–5322. [Google Scholar] [CrossRef]
- Han, M.V.; Thomas, G.W.; Lugo-Martinez, J.; Hahn, M.W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef]
- Sun, P.; Jiao, B.; Yang, Y.; Shan, L.; Li, T.; Li, X.; Xi, Z.; Wang, X.; Liu, J. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Briere, C.; Owens, G.L.; Carrere, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Krishnakumar, V.; Li, J.; jcvi: JCVI Utility Libraries. Zenodo. 2015. Available online: https://zenodo.org/records/31631 (accessed on 22 January 2023).
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2021, 50, D387–D390. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Genome size | 1.55 Gb |
| Contig N50 | 25.66 Mb |
| Scaffold N50 | 58.10 Mb |
| Anchored to chromosome | 1.52 Gb (97.79%) |
| GC content | 33.77% |
| Number of chromosomes | 26 |
| Repetitive sequences | 1.15 Gb (77.81%) |
| Number of protein-coding genes | 26,048 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, F.; Li, B.; Song, K.; Wang, Y.; Hou, Z.; Li, H.; Zhang, X.; Li, F.; Yang, L. Polyploid Genome Assembly Provides Insights into Morphological Development and Ascorbic Acid Accumulation of Sauropus androgynus. Int. J. Mol. Sci. 2024, 25, 300. https://doi.org/10.3390/ijms25010300
Xia F, Li B, Song K, Wang Y, Hou Z, Li H, Zhang X, Li F, Yang L. Polyploid Genome Assembly Provides Insights into Morphological Development and Ascorbic Acid Accumulation of Sauropus androgynus. International Journal of Molecular Sciences. 2024; 25(1):300. https://doi.org/10.3390/ijms25010300
Chicago/Turabian StyleXia, Fagang, Bin Li, Kangkang Song, Yankun Wang, Zhuangwei Hou, Haozhen Li, Xiaohua Zhang, Fangping Li, and Long Yang. 2024. "Polyploid Genome Assembly Provides Insights into Morphological Development and Ascorbic Acid Accumulation of Sauropus androgynus" International Journal of Molecular Sciences 25, no. 1: 300. https://doi.org/10.3390/ijms25010300