
Tissue Sodium Accumulation Induces Organ Inflammation and Injury in Chronic Kidney Disease
 , and
, and
Abstract
:1. Introduction
2. Sodium Intake and Storage in CKD Patients and Healthy Individuals
3. TonEBP Activation with and without Sodium Storage in Tissues
4. Salt-Induced Tissue Inflammation in Renal Failure Model Mice with Salt Loading
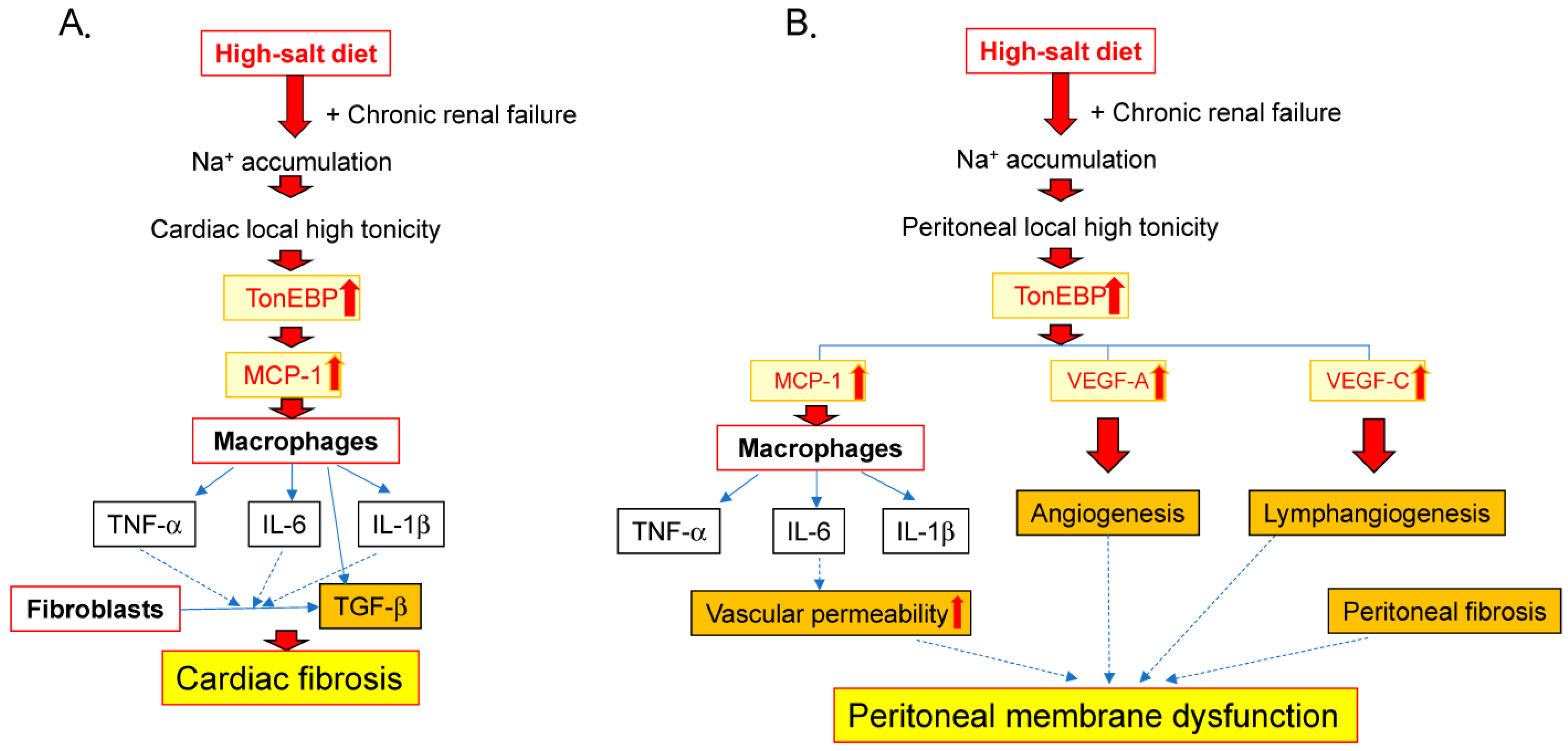
5. Salt-Induced Cardiac Inflammation and Fibrosis in Renal Failure
6. Salt-Induced Inflammation in the Peritoneum during Renal Failure
7. Strategy to Ameliorate Inflammation Induced by Sodium Storage-Related TonEBP Stimulation in Renal Failure

{kind=link}
{kind=link}
{kind=link}
| Human/ Mouse | Number of Subjects | Agent | Outcome | Ref. |
|---|---|---|---|---|
| human | 22 maintenance HD patients | Anakinra (IL-1 receptor antagonist) | Significant increase in serum CRP and IL-6 levels | [152] |
| human | 22 maintenance HD patients | Anakinra (IL-1 receptor antagonist) | Significant increase in serum adiponectin levels | [157] |
| human | 42 CKD stage 3 and 4 patients | Rilonacept (IL-1 trap) | Significant decrease in brachial artery FMD, the assessment of endothelial function | [154] |
| human | 61 maintenance HD patients | Ziltivekimab (anti–IL-6 antibody) | Significant increase in serum albumin and decrease in ERI and ESA usage | [151] |
| human | 264 maintenance HD patients | Ziltivekimab (anti–IL-6 antibody) | Significant decrease in serum hsCRP, fibrinogen, haptoglobin, SAA, sPLA2, lipoprotein (a), | [108] |
| mouse | 32 mice with acute kidney injury after IRI | Etanercept (TNF-α antibody) | Significant decrease in kidney fibrosis of about 25% and blockade of AKI-to-CKD transition | [158] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CKD | chronic kidney disease |
| ESL | endothelial surface layer |
| eGFR | estimated glomerular filtration rate |
| GAG | glycosaminoglycan |
| IL | interleukin |
| MCP-1 | monocyte chemoattractant protein-1 |
| miRNA | microRNA |
| MRI | magnetic resonance imaging |
| Nx | subtotal (5/6) nephrectomy |
| Nx-rat IgG1 | rat IgG1-treated Nx mice with salt loading |
| Nx-MR16-1 | MR16-1-treated Nx mice with salt loading |
| PD | peritoneal dialysis |
| PSTR | peritoneal solute transport rate |
| Sgk1 | serum and glucocorticoid-regulated kinase 1 |
| 5/6Nx | sub-total-nephrectomized mice |
| 5/6Nx/NaCl | 5/6Nx with 1% NaCl loading |
| 5/6Nx/Water | 5/6Nx with tap water administration |
| Sham/Water | sham surgery with tap water |
| TGF-β | transforming growth factor-beta |
| TNF-α | tumor necrosis factor-alpha |
| TonEBP | tonicity-responsive enhancer-binding protein |
| UACR | urinary albumin-to-creatinine ratio |
| VEGF-A | vascular endothelial growth factor A |
| VEGF-C | vascular endothelial growth factor C |
References
- Borrelli, S.; De Nicola, L.; De Gregorio, I.; Polese, L.; Pennino, L.; Elefante, C.; Carbone, A.; Rappa, T.; Minutolo, R.; Garofalo, C. Volume-Independent Sodium Toxicity in Peritoneal Dialysis: New Insights from Bench to Bed. Int. J. Mol. Sci. 2021, 22, 12804. [Google Scholar] [CrossRef] [PubMed]
- Titze, J.; Luft, F.C. Speculations on salt and the genesis of arterial hypertension. Kidney Int. 2017, 91, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Kawaguchi, Y. Multicenter survey on hydration status and control of blood pressure in Japanese CAPD patients. Perit. Dial. Int. 2002, 22, 411–414. [Google Scholar] [PubMed]
- Valtuille, R. Potential Novel Benefits of Sodium Restriction in Chronic Kidney Disease. Curr. Hypertens. Rev. 2021, 17, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, S.; Luft, F.C.; Titze, J.; Kitada, K. Sodium Handling and Interaction in Numerous Organs. Am. J. Hypertens. 2020, 33, 687–694. [Google Scholar] [CrossRef]
- Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Temmar, M.; Lemke, H.D.; Tribouilloy, C.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. 2010, 77, 550–556. [Google Scholar] [CrossRef]
- Menon, V.; Greene, T.; Wang, X.; Pereira, A.A.; Marcovina, S.M.; Beck, G.J.; Kusek, J.W.; Collins, A.J.; Levey, A.S.; Sarnak, M.J. C-reactive protein and albumin as predictors of all-cause and cardiovascular mortality in chronic kidney disease. Kidney Int. 2005, 68, 766–772. [Google Scholar] [CrossRef]
- Takahashi, R.; Ito, Y.; Takahashi, H.; Ishii, H.; Kasuga, H.; Mizuno, M.; Suzuki, Y.; Yuzawa, Y.; Maruyama, S.; Murohara, T.; et al. Combined values of serum albumin, C-reactive protein and body mass index at dialysis initiation accurately predicts long-term mortality. Am. J. Nephrol. 2012, 36, 136–143. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Gillespie, I.A.; Tunks, J.; Addison, J.; Kronenberg, F.; Drueke, T.B.; Marcelli, D.; Schernthaner, G.; Eckardt, K.; Floege, J.; et al. Inflammation Modifies the Paradoxical Association between Body Mass Index and Mortality in Hemodialysis Patients. J. Am. Soc. Nephrol. 2016, 27, 1479–1486. [Google Scholar] [CrossRef]
- Fouque, D.; Kalantar-Zadeh, K.; Kopple, J.; Cano, N.; Chauveau, P.; Cuppari, L.; Franch, H.; Guarnieri, G.; Ikizler, T.A.; Kaysen, G.; et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int. 2008, 73, 391–398. [Google Scholar] [CrossRef]
- Honda, H.; Qureshi, A.R.; Heimbürger, O.; Barany, P.; Wang, K.; Pecoits-Filho, R.; Stenvinkel, P.; Lindholm, B. Serum albumin, C-reactive protein, interleukin 6, and fetuin a as predictors of malnutrition, cardiovascular disease, and mortality in patients with ESRD. Am. J. Kidney Dis. 2006, 47, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Herzog, C.A. Cardiovascular disease in chronic kidney disease. In Comprehensive Clinical Nephrology; Johnson, R., Feehally, J., Floege, J., Tonelli, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 942–957. [Google Scholar]
- Borrelli, S.; Provenzano, M.; Gagliardi, I.; Michael, A.; Liberti, M.E.; De Nicola, L.; Conte, G.; Garofalo, C.; Andreucci, M. Sodium Intake and Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4744. [Google Scholar] [CrossRef]
- Koomans, H.A.; Roos, J.C.; Dorhout Mees, E.J.; Delawi, I.M. Sodium balance in renal failure. A comparison of patients with normal subjects under extremes of sodium intake. Hypertension 1985, 7, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.T.; Su, G.; Zhang, L.; Qin, X.; Marshall, S.; González-Ortiz, A.; Clase, C.M.; Campbell, K.L.; Xu, H.; Carrero, J.J. Modifiable Lifestyle Factors for Primary Prevention of CKD: A Systematic Review and Meta-Analysis. J. Am. Soc. Nephrol. 2021, 32, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Izumaru, K.; Hata, J.; Nakano, T.; Nakashima, Y.; Nagata, M.; Fukuhara, M.; Oda, Y.; Kitazono, T.; Ninomiya, T. Reduced Estimated GFR and Cardiac Remodeling: A Population-Based Autopsy Study. Am. J. Kidney Dis. 2019, 74, 373–381. [Google Scholar] [CrossRef]
- Borrelli, S.; Mallamaci, F.; Chiodini, P.; Garofalo, C.; Pizzini, P.; Tripepi, R.; D’Arrigo, G.; Tripepi, G.; Conte, G.; Nicola, L.D.; et al. Salt intake correlates with night systolic blood pressure in non-dialytic chronic kidney disease. Nephrol. Dial. Transplant. 2022, 37, 1387–1389. [Google Scholar] [CrossRef]
- Shi, H.; Su, X.; Li, C.; Guo, W.; Wang, L. Effect of a low-salt diet on chronic kidney disease outcomes: A systematic review and meta-analysis. BMJ Open. 2022, 12, e050843. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef]
- Ayuzawa, N.; Fujita, T. The Mineralocorticoid Receptor in Salt-Sensitive Hypertension and Renal Injury. J. Am. Soc. Nephrol. 2021, 32, 279–289. [Google Scholar] [CrossRef]
- Meuleman, Y.; Hoekstra, T.; Dekker, F.W.; Navis, G.; Vogt, L.; van der Boog, P.J.M.; Bos, W.J.; van Montfrans, G.A.; van Dijk, S.; ESMO Study Group. Sodium Restriction in Patients with CKD: A Randomized Controlled Trial of Self-management Support. Am. J. Kidney. Dis. 2017, 69, 576–586. [Google Scholar] [CrossRef]
- Mills, K.T.; Chen, J.; Yang, W.; Appel, L.J.; Kusek, J.W.; Alper, A.; Delafontaine, P.; Keane, M.G.; Mohler, E.; Ojo, A.; et al. Sodium Excretion and the Risk of Cardiovascular Disease in Patients With Chronic Kidney Disease. JAMA 2016, 315, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Chan, Q.; Daviglus, M.L.; Dyer, A.R.; Van Horn, L.; Garside, D.B.; Garside, D.B.; Miura, M.; Wu, Y.; Ueshima, H.; et al. Relation of Dietary Sodium (Salt) to Blood Pressure and Its Possible Modulation by Other Dietary Factors: The INTERMAP Study. Hypertension 2018, 71, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Chang, T.I.; Cushman, W.C.; Furth, S.L.; Hou, F.F.; Ix, J.H.; Knoll, G.A.; Muntner, P.; Pecoits-Filho, R.; Sarnak, M.J.; et al. Executive summary of the KDIGO 2021 Clinical Practice Guideline for the Management of Blood Pressure in Chronic Kidney Disease. Kidney Int. 2021, 99, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Wu, Y.; Feng, X.; Zhang, R.; Zhang, Y.; Shi, J.; Zhang, J.; Tian, M.; Huang, L.; Li, Z.; et al. Effect of Salt Substitution on Cardiovascular Events and Death. N. Engl. J. Med. 2021, 385, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Galletti, F.; Agabiti-Rosei, E.; Bernini, G.; Boero, R.; Desideri, G.; Fallo, F.; Mallamaci, F.; Morganti, A.; Castellano, M.; Nazzaro, P.; et al. Excess dietary sodium and inadequate potassium intake by hypertensive patients in Italy: Results of the MINISAL-SIIA study program. J. Hypertens. 2014, 32, 48–56. [Google Scholar] [CrossRef]
- Rakova, N.; Jüttner, K.; Dahlmann, A.; Schröder, A.; Linz, P.; Kopp, C.; Rauh, M.; Goller, U.; Beck, L.; Agureev, A.; et al. Long-term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell. Metab. 2013, 17, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Cuzzola, F.; Mallamaci, F.; Tripepi, G.; Parlongo, S.; Cutrupi, S.; Cataliotti, A.; Stancanelli, B.; Malatino, L.; Bellanuova, I.; Ferri, C.; et al. Urinary adrenomedullin is related to ET-1 and salt intake in patients with mild essential hypertension. Salt Sensitivity Group of Italian Society of Hypertension. Am. J. Hypertens. 2001, 14, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Malatino, L.S.; Bellanuova, I.; Cataliotti, A.; Cuzzola, F.; Mallamaci, F.; Tripepi, G.; Parlongo, S.; Cutrupi, S.; Mangiafico, R.A.; Ferri, C.; et al. Renal endothelin-1 is linked to changes in urinary salt and volume in essential hypertension. Salt Sensitivity Group of the Italian Society of Hypertension. J. Nephrol. 2000, 13, 178–184. [Google Scholar]
- Kopp, C.; Linz, P.; Wachsmuth, L.; Dahlmann, A.; Horbach, T.; Schöfl, C.; Renz, W.; Santoro, D.; Niendorf, T.; Müller, D.N.; et al. 23Na magnetic resonance imaging of tissue sodium. Hypertension 2012, 59, 167–172. [Google Scholar] [CrossRef]
- Kopp, C.; Linz, P.; Dahlmann, A.; Hammon, M.; Jantsch, J.; Müller, D.N.; Schmieder, R.E.; Cavallaro, A.; Eckardt, K.U.; Uder, M.; et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 2013, 61, 635–640. [Google Scholar] [CrossRef]
- Dahlmann, A.; Dörfelt, K.; Eicher, F.; Linz, P.; Kopp, C.; Mössinger, I.; Horn, S.; Büschges-Seraphin, B.; Wabel, P.; Hammon, M.; et al. Magnetic resonance-determined sodium removal from tissue stores in hemodialysis patients. Kidney Int. 2015, 87, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.P.; Raff, U.; Kopp, C.; Scheppach, J.B.; Toncar, S.; Wanner, C.; Schlieper, G.; Saritas, T.; Floege, J.; Schmid, M.; et al. Skin Sodium Concentration Correlates with Left Ventricular Hypertrophy in CKD. J. Am. Soc. Nephrol. 2017, 28, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Mitsides, N.; Alsehli, F.M.S.; Mc Hough, D.; Shalamanova, L.; Wilkinson, F.; Alderdice, J.; Mitra, R.; Swiecicka, A.; Brenchley, P.; Parker, G.J.M.; et al. Salt and Water Retention Is Associated with Microinflammation and Endothelial Injury in Chronic Kidney Disease. Nephron 2019, 143, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Mitsides, N.; McHugh, D.; Swiecicka, A.; Mitra, R.; Brenchley, P.; Parker, G.J.M.; Mitra, S. Extracellular resistance is sensitive to tissue sodium status; implications for bioimpedance-derived fluid volume parameters in chronic kidney disease. J. Nephrol. 2020, 33, 119–127. [Google Scholar] [CrossRef]
- Qirjazi, E.; Salerno, F.R.; Akbari, A.; Hur, L.; Penny, J.; Scholl, T.; Scholl, T.; McIntyre, C.W. Tissue sodium concentrations in chronic kidney disease and dialysis patients by lower leg sodium-23 magnetic resonance imaging. Nephrol. Dial. Transplant. 2020, 36, 1234–1243. [Google Scholar] [CrossRef]
- Sahinoz, M.; Tintara, S.; Deger, S.M.; Alsouqi, A.; Crescenzi, R.L.; Mambungu, C.; Vincz, A.; Mason, O.; Prigmore, H.L.; Guide, A.; et al. Tissue sodium stores in peritoneal dialysis and hemodialysis patients determined by 23-sodium magnetic resonance imaging. Nephrol. Dial. Transplant. 2020, 36, 1307–1317. [Google Scholar] [CrossRef]
- Lemoine, S.; Salerno, F.R.; Akbari, A.; McKelvie, R.S.; McIntyre, C.W. Tissue Sodium Storage in Patients with Heart Failure: A New Therapeutic Target? Circ. Cardiovasc. Imaging 2021, 14, e012910. [Google Scholar] [CrossRef]
- Friedrich, A.C.; Linz, P.; Nagel, A.M.; Rosenhauer, D.; Horn, S.; Schiffer, M.; Uder, M.; Kopp, C.; Dahlmann, A. Hemodialysis Patients with Cardiovascular Disease Reveal Increased Tissue Na+ Deposition. Kidney Blood Press. Res. 2022, 47, 185–193. [Google Scholar] [CrossRef]
- Salerno, F.R.; Akbari, A.; Lemoine, S.; Scholl, T.J.; McIntyre, C.W.; Filler, G. Effects of pediatric chronic kidney disease and its etiology on tissue sodium concentration: A pilot study. Pediatr. Nephrol. 2023, 38, 499–507. [Google Scholar] [CrossRef]
- Salerno, F.R.; Akbari, A.; Lemoine, S.; Filler, G.; Scholl, T.J.; McIntyre, C.W. Outcomes and predictors of skin sodium concentration in dialysis patients. Clin. Kidney J. 2022, 15, 1129–1136. [Google Scholar] [CrossRef]
- Neuhofer, W. Role of NFAT5 in inflammatory disorders associated with osmotic stress. Curr. Genomics. 2010, 11, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Ferraris, J.D.; Dmitrieva, N.I. Cellular response to hyperosmotic stresses. Physiol. Rev. 2007, 87, 1441–1474. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lee-Kwon, W.; Kwon, H.M. The evolving role of TonEBP as an immunometabolic stress protein. Nat. Rev. Nephrol. 2020, 16, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Aghakeshmiri, S.; Chen, Y.T.; Zhang, H.M.; Yip, F.; Yang, D. NFAT5-Mediated Signalling Pathways in Viral Infection and Cardiovascular Dysfunction. Int. J. Mol. Sci. 2021, 22, 4872. [Google Scholar] [CrossRef]
- Yamauchi, A.; Uchida, S.; Kwon, H.M.; Preston, A.S.; Robey, R.B.; Garcia-Perez, A.; Burg, M.B.; Handleret, J.S. Cloning of a Na(+)- and Cl(-)-dependent betaine transporter that is regulated by hypertonicity. J. Biol. Chem. 1992, 267, 649–652. [Google Scholar] [CrossRef]
- Burg, M.B.; Kwon, E.D.; Kültz, D. Regulation of gene expression by hypertonicity. Annu. Rev. Physiol. 1997, 59, 437–455. [Google Scholar] [CrossRef]
- Bauernschmitt, H.G.; Kinne, R.K. Metabolism of the ‘organic osmolyte’ glycerophosphorylcholine in isolated rat inner medullary collecting duct cells. I. Pathways for synthesis and degradation. Biochim. Biophys. Acta 1993, 1148, 331–341. [Google Scholar] [CrossRef]
- Bauernschmitt, H.G.; Kinne, R.K. Metabolism of the ‘organic osmolyte’ glycerophosphorylcholine in isolated rat inner medullary collecting duct cells. II. Regulation by extracellular osmolality. Biochim. Biophys. Acta 1993, 1150, 25–34. [Google Scholar] [CrossRef]
- Sakata, F.; Ito, Y.; Mizuno, M.; Sawai, A.; Suzuki, Y.; Tomita, T.; Tawada, M.; Tanaka, A.; Hirayama, A.; Sagaraet, A.; et al. Sodium chloride promotes tissue inflammation via osmotic stimuli in subtotal-nephrectomized mice. Lab. Investig. 2017, 97, 432–446. [Google Scholar] [CrossRef]
- Arnold, C.; Feldner, A.; Zappe, M.; Komljenovic, D.; De La Torre, C.; Ruzicka, P.; Hecker, M.; Neuhofer, W.; Korff, T. Genetic ablation of NFAT5/TonEBP in smooth muscle cells impairs flow- and pressure-induced arterial remodeling in mice. FASEB J. 2019, 33, 3364–3377. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lim, S.W.; Salimi, S.; Yoo, E.J.; Lee-Kwon, W.; Lee, H.H.; Mitchell, B.D.; Sanada, S.; Parsa, A.; Kwon, H.M. Tonicity-Responsive Enhancer-Binding Protein Mediates Hyperglycemia-Induced Inflammation and Vascular and Renal Injury. J. Am. Soc. Nephrol. 2018, 29, 492–504. [Google Scholar] [CrossRef]
- Lee, H.H.; Sanada, S.; An, S.M.; Ye, B.J.; Lee, J.H.; Seo, Y.K.; Lee, C.; Lee-Kwon, W.; Küper, C.; Neuhofer, W.; et al. LPS-induced NFκB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci. Rep. 2016, 6, 24921. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, J.; López-Rodríguez, C. Regulation of Inflammatory Functions of Macrophages and T Lymphocytes by NFAT5. Front. Immunol. 2019, 10, 535. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kong, L.B.; Li, J.T.; Guo, Z.Y.; Xue, Q.; Yang, T.; Meng, M.; Jin, G.Q.; Wen, W.H.; Yang, A.G. MiR-568 inhibits the activation and function of CD4⁺ T cells and Treg cells by targeting NFAT5. Int. Immunol. 2014, 26, 269–281. [Google Scholar] [CrossRef]
- Ying, W.; Tseng, A.; Chang, R.C.; Morin, A.; Brehm, T.; Triff, K.; Nair, V.; Zhuang, G.; Song, H.; Kanameni, S.; et al. MicroRNA-223 is a crucial mediator of PPARγ-regulated alternative macrophage activation. J. Clin. Investig. 2015, 125, 4149–4159. [Google Scholar] [CrossRef]
- Xin, Y.; Cai, H.; Lu, T.; Zhang, Y.; Yang, Y.; Cui, Y. miR-20b Inhibits T Cell Proliferation and Activation via NFAT Signaling Pathway in Thymoma-Associated Myasthenia Gravis. Biomed. Res. Int. 2016, 2016, 9595718. [Google Scholar] [CrossRef]
- Hinske, L.C.; Heyn, J.; Hübner, M.; Rink, J.; Hirschberger, S.; Kreth, S. Intronic miRNA-641 controls its host Gene’s pathway PI3K/AKT and this relationship is dysfunctional in glioblastoma multiforme. Biochem. Biophys. Res. Commun. 2017, 489, 477–483. [Google Scholar] [CrossRef]
- Tao, H.; Xiong, Q.; Ji, Z.; Zhang, F.; Liu, Y.; Chen, M. NFAT5 is Regulated by p53/miR-27a Signal Axis and Promotes Mouse Ovarian Granulosa Cells Proliferation. Int. J. Biol. Sci. 2019, 15, 287–297. [Google Scholar] [CrossRef]
- Meng, X.; Li, Z.; Zhou, S.; Xiao, S.; Yu, P. miR-194 suppresses high glucose-induced non-small cell lung cancer cell progression by targeting NFAT5. Thorac. Cancer 2019, 10, 1051–1059. [Google Scholar] [CrossRef]
- Ge, G.; Yang, D.; Tan, Y.; Chen, Y.; Jiang, D.; Jiang, A.; Li, Q.; Liu, Y.; Zhong, Z.; Li, X.; et al. miR-10b-5p Regulates C2C12 Myoblasts Proliferation and Differentiation. Biosci. Biotechnol. Biochem. 2019, 83, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; You, S.; Yoo, S.A.; Kim, N.H.; Kwon, H.M.; Yoon, C.H.; Cho, C.S.; Hwang, D.; Kim, W.U. NF-AT5 is a critical regulator of inflammatory arthritis. Arthritis Rheum. 2011, 63, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kong, J.S.; You, S.; Kwon, H.M.; Yoo, S.A.; Cho, C.S.; Kim, W.U. Transcription Factor NFAT5 Promotes Migration and Invasion of Rheumatoid Synoviocytes via Coagulation Factor III and CCL2. J. Immunol. 2018, 201, 359–370. [Google Scholar] [CrossRef] [PubMed]
- López-Rodríguez, C.; Antos, C.L.; Shelton, J.M.; Richardson, J.A.; Lin, F.; Novobrantseva, T.I.; Bronson, R.T.; Igarashi, P.; Rao, A.; Olson, E.N. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 2392–2397. [Google Scholar] [CrossRef]
- Berry, M.R.; Mathews, R.J.; Ferdinand, J.R.; Jing, C.; Loudon, K.W.; Wlodek, E.; Dennison, T.W.; Kuper, C.; Neuhofer, W.; Clatworthy, M.R. Renal Sodium Gradient Orchestrates a Dynamic Antibacterial Defense Zone. Cell 2017, 170, 860–874.e19. [Google Scholar] [CrossRef]
- Jantsch, J.; Schatz, V.; Friedrich, D.; Schröder, A.; Kopp, C.; Siegert, I.; Maronna, A.; Wendelborn, D.; Linz, P.L.; Binger, K.J.; et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015, 21, 493–501. [Google Scholar] [CrossRef]
- Berga-Bolaños, R.; Drews-Elger, K.; Aramburu, J.; López-Rodríguez, C. NFAT5 regulates T lymphocyte homeostasis and CD24-dependent T cell expansion under pathologic hypernatremia. J. Immunol. 2010, 185, 6624–6635. [Google Scholar] [CrossRef]
- Neubert, P.; Weichselbaum, A.; Reitinger, C.; Schatz, V.; Schröder, A.; Ferdinand, J.R.; Simon, M.; Bär, A.L.; Brochhausen, C.; Gerlach, R.G.; et al. HIF1A and NFAT5 coordinate Na(+)-boosted antibacterial defense via enhanced autophagy and autolysosomal targeting. Autophagy 2019, 15, 1899–1916. [Google Scholar] [CrossRef]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.K.; Beck, F.X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Moriguchi, Y.; Yogo, K.; Aizawa, K.; Serizawa, K.; Tashiro, Y.; Yorozu, K.; Ishizuka, N.; Iwabuchi, S.; Kitamura, H.; Nishimura, T. Left ventricular hypertrophy is associated with inflammation in sodium loaded subtotal nephrectomized rats. Biomed. Res. 2011, 32, 83–90. [Google Scholar] [CrossRef]
- Van Koppen, A.; Verhaar, M.C.; Bongartz, L.G.; Joles, J.A. 5/6th nephrectomy in combination with high salt diet and nitric oxide synthase inhibition to induce chronic kidney disease in the Lewis rat. J. Vis. Exp. 2013, 77, e50398. [Google Scholar]
- Cao, W.; Li, A.; Wang, L.; Zhou, Z.; Su, Z.; Bin, W.; Wilcox, C.S.; Hou, F.F. A Salt-Induced Reno-Cerebral Reflex Activates Renin-Angiotensin Systems and Promotes CKD Progression. J. Am. Soc. Nephrol. 2015, 26, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Sakata, F.; Ishii, T.; Tawada, M.; Suzuki, Y.; Kinashi, H.; Katsuno, T.; Takei, Y.; Maruyama, S.; Mizuno, M.; et al. Excessive salt intake increases peritoneal solute transport rate via local tonicity-responsive enhancer binding protein in subtotal nephrectomized mice. Nephrol. Dial. Transplant. 2019, 34, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, K.C.; Zambom, F.F.F.; Albino, A.H.; Alarcon Arias, S.C.; Ávila, V.F.; Faustino, V.D.; Malheiros, D.M.A.C.; Camara, N.O.S.; Fujihara, C.K.; Zatz, R. NF-κB blockade during short-term l-NAME and salt overload strongly attenuates the late development of chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2020, 319, F215–F228. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, M.; Zeng, T.; Qiu, M.; Zhang, M.; Jiang, S.; Tan, L.; Li, A. Silencing of Central (Pro)renin Receptor Ameliorates Salt-Induced Renal Injury in Chronic Kidney Disease. Antioxid. Redox. Signal. 2021, 35, 93–112. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, X.; Yang, S.; Peng, Y.; Hou, F.; Zhou, Q. High salt aggravates renal inflammation via promoting pro-inflammatory macrophage in 5/6-nephrectomized rat. Life Sci. 2021, 274, 119109. [Google Scholar] [CrossRef]
- Tanaka, H.; Sun, T.; Kinashi, H.; Kamiya, K.; Yamaguchi, M.; Nobata, H.; Sakata, F.; Kim, H.; Mizuno, M.; Kunoki, S.; et al. Interleukin-6 blockade reduces salt-induced cardiac inflammation and fibrosis in subtotal nephrectomized mice. Am. J. Physiol. Renal Physiol. 2022, 323, F654–F665. [Google Scholar] [CrossRef]
- Kottgen, A.; Russell, S.D.; Loehr, L.R.; Crainiceanu, C.M.; Rosamond, W.D.; Chang, P.P.; Chambless, L.E.; Coresh, J. Reduced kidney function as a risk factor for incident heart failure: The atherosclerosis risk in communities (ARIC) study. J. Am. Soc. Nephrol. 2007, 18, 1307–1315. [Google Scholar] [CrossRef]
- Matsushita, K.; Ballew, S.H.; Wang, A.Y.; Kalyesubula, R.; Schaeffner, E.; Agarwal, R. Epidemiology and risk of cardiovascular disease in populations with chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 696–707. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease. J. Clin. Investig. 2007, 117, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Voloshenyuk, T.G.; Hart, A.D.; Khoutorova, E.; Gardner, J.D. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem. Biophys. Res. Commun. 2011, 413, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Park, S.J.; Hwang, H.Y.; Park, E.J.; Nam, J.H.; Kim, J.; Park, S.I. TGF-beta1 induces cardiac hypertrophic responses via PKC-dependent ATF-2 activation. J. Mol. Cell. Cardiol. 2005, 39, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, F.J.; Dillmann, W.H. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am. J. Physiol. 1992, 262, H1861–H1866. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Querejeta, R.; López, B.; González, A.; Sánchez, E.; Larman, M.; Martínez Ubago, J.L.; Díez, J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef]
- Löfman, I.; Szummer, K.; Dahlström, U.; Jernberg, T.; Lund, L.H. Associations with and prognostic impact of chronic kidney disease in heart failure with preserved, mid-range, and reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1606–1614. [Google Scholar] [CrossRef]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Gheorghiade, M.; Bonow, R.O.; Huang, C.C.; Deo, R.C. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.H.; Cheng, P.Y.; Shih, N.L.; Chen, I.B.; Wang, D.L.; Chen, J.J. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J. Am. Coll. Cardiol. 2003, 42, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.; Papparella, I.; Petrov, V.; Semplicini, A.; Fagard, R. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J. Hypertens. 2006, 24, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef]
- Glassock, R.J.; Pecoits-Filho, R.; Barberato, S.H. Left ventricular mass in chronic kidney disease and ESRD. Clin. J. Am. Soc. Nephrol. 2009, 4 (Suppl. 1), S79–S91. [Google Scholar] [CrossRef]
- Romero-González, G.; González, A.; López, B.; Ravassa, S.; Díez, J. Heart failure in chronic kidney disease: The emerging role of myocardial fibrosis. Nephrol. Dial. Transplant. 2022, 37, 817–824. [Google Scholar] [CrossRef]
- Wang, X.; Shapiro, J.I. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat. Rev. Nephrol. 2019, 15, 159–175. [Google Scholar] [CrossRef]
- Baldo, M.P.; Rodrigues, S.L.; Mill, J.G. High salt intake as a multifaceted cardiovascular disease: New support from cellular and molecular evidence. Heart Fail. Rev. 2015, 20, 461–474. [Google Scholar] [CrossRef]
- Takahashi, H.; Ishii, H.; Aoyama, T.; Kamoi, D.; Kasuga, H.; Ito, Y.; Yasuda, K.; Tanaka, M.; Yoshikawa, D.; Maruyama, S.; et al. Association of cardiac valvular calcifications and C-reactive protein with cardiovascular mortality in incident hemodialysis patients: A Japanese cohort study. Am. J. Kidney Dis. 2013, 61, 254–261. [Google Scholar] [CrossRef]
- Takahashi, H.; Ito, Y.; Ishii, H.; Aoyama, T.; Kamoi, D.; Kasuga, H.; Yasuda, K.; Maruyama, S.; Matsuo, S.; Murohara, T.; et al. Geriatric nutritional risk index accurately predicts cardiovascular mortality in incident hemodialysis patients. J. Cardiol. 2014, 64, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zheng, J.; Miao, Y.; Wang, Y.; Cui, W.; Guo, J.; Qiu, S.; Han, Y.; Jia, L.; Li, H.; et al. Serum-glucocorticoid regulated kinase 1 regulates alternatively activated macrophage polarization contributing to angiotensin II-induced inflammation and cardiac fibrosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Aiba, T.; Rosenberg, M.; Hessler, K.; Xiao, C.; Quintero, P.A.; Ottaviano, F.G.; Knight, A.C.; Graham, E.L.; Boström, P.; et al. Pathological role of serum- and glucocorticoid-regulated kinase 1 in adverse ventricular remodeling. Circulation 2012, 126, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- López-Rodríguez, C.; Aramburu, J.; Jin, L.; Rakeman, A.S.; Michino, M.; Rao, A. Bridging the NFAT and NF-kappaB families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity 2001, 15, 47–58. [Google Scholar]
- Mak, M.C.; Lam, K.M.; Chan, P.K.; Lau, Y.B.; Tang, W.H.; Yeung, P.K.; Ko, B.C.; Chung, S.M.; Chung, S.K. Embryonic lethality in mice lacking the nuclear factor of activated T cells 5 protein due to impaired cardiac development and function. PLoS ONE 2011, 6, e19186. [Google Scholar] [CrossRef]
- Küper, C.; Beck, F.X.; Neuhofer, W. Generation of a conditional knockout allele for the NFAT5 gene in mice. Front. Physiol. 2014, 5, 507. [Google Scholar]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Lott, J.; Platt, T.; Niesporek, S.C.; Paech, D.; Behl, N.G.R.; Niendorf, T.; Bachert, P.; Ladd, M.E.; Nagel, A.M. Corrections of myocardial tissue sodium concentration measurements in human cardiac (23) Na MRI at 7 Tesla. Magn. Reson. Med. 2019, 82, 159–173. [Google Scholar] [CrossRef]
- Li, P.K.; Chow, K.M.; Van de Luijtgaarden, M.W.; Johnson, D.W.; Jager, K.J.; Mehrotra, R.; Naicker, S.; Pecoits-Filho, R.; Yu, X.Q.; Lameire, N. Changes in the worldwide epidemiology of peritoneal dialysis. Nat. Rev. Nephrol. 2017, 13, 90–103. [Google Scholar] [CrossRef]
- Borrelli, S.; La Milia, V.; De Nicola, L.; Cabiddu, G.; Russo, R.; Provenzano, M.; Minutolo, R.; Conte, G.; Garofalo, C. Sodium removal by peritoneal dialysis: A systematic review and meta-analysis. J. Nephrol. 2019, 32, 231–239. [Google Scholar] [CrossRef]
- Pletinck, A.; Vanholder, R.; Veys, N.; Van Biesen, W. Protecting the peritoneal membrane: Factors beyond peritoneal dialysis solutions. Nat. Rev. Nephrol. 2012, 8, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Brimble, K.S.; Walker, M.; Margetts, P.J.; Kundhal, K.K.; Rabbat, C.G. Meta-analysis: Peritoneal membrane transport, mortality, and technique failure in peritoneal dialysis. J. Am. Soc. Nephrol. 2006, 17, 2591–2598. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.H.; Jung, J.Y.; Yoon, M.O.; Song, A.; Lee, H.; Ro, H.; Hwang, Y.H.; Kim, D.K.; Margetts, P.; Ahn, C. Intra-peritoneal interleukin-6 system is a potent determinant of the baseline peritoneal solute transport in incident peritoneal dialysis patients. Nephrol. Dial. Transpl. 2010, 25, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Pecoits-Filho, R.; Carvalho, M.J.; Stenvinkel, P.; Lindholm, B.; Heimbürger, O. Systemic and intraperitoneal interleukin-6 system during the first year of peritoneal dialysis. Perit. Dial. Int. 2006, 26, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Sawai, A.; Ito, Y.; Mizuno, M.; Suzuki, Y.; Toda, S.; Ito, I.; Hattori, R.; Matsukawa, Y.; Gotoh, M.; Takei, Y.; et al. Peritoneal macrophage infiltration is correlated with baseline peritoneal solute transport rate in peritoneal dialysis patients. Nephrol. Dial. Transpl. 2011, 26, 2322–2332. [Google Scholar] [CrossRef]
- Pletinck, A.; Consoli, C.; Van Landschoot, M.; Steppan, S.; Topley, N.; Passlick-Deetjen, J.; Vanholder, R.; Van Biesen, W. Salt intake induces epithelial-to-mesenchymal transition of the peritoneal membrane in rats. Nephrol. Dial. Transpl. 2010, 25, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Kitterer, D.; Latus, J.; Ulmer, C.; Fritz, P.; Biegger, D.; Ott, G.; Alscher, M.D.; Witowski, J.; Kawka, E.; Jörres, A.; et al. Activation of nuclear factor of activated T cells 5 in the peritoneal membrane of uremic patients. Am. J. Physiol. Renal Physiol. 2015, 308, F1247–F1258. [Google Scholar] [CrossRef]
- Küper, C.; Beck, F.X.; Neuhofer, W. NFAT5 contributes to osmolality-induced MCP-1 expression in mesothelial cells. Mediat. Inflamm. 2012, 2012, 513015. [Google Scholar] [CrossRef]
- Rodrigues-Díez, R.; Aroeira, L.S.; Orejudo, M.; Bajo, M.A.; Heffernan, J.J.; Rodrigues-Díez, R.R.; Rayego-Mateos, S.; Ortiz, A.; Gonzalez-Mateo, G.; López-Cabrera, M.; et al. IL-17A is a novel player in dialysis-induced peritoneal damage. Kidney Int. 2014, 86, 303–315. [Google Scholar] [CrossRef]
- Titze, J.; Lang, R.; Ilies, C.; Schwind, K.H.; Kirsch, K.A.; Dietsch, P.; Luft, F.C.; Hilgers, K.F. Osmotically inactive skin Na+ storage in rats. Am. J. Physiol. Renal Physiol. 2003, 285, F1108–F1117. [Google Scholar] [CrossRef]
- Titze, J.; Shakibaei, M.; Schafflhuber, M.; Schulze-Tanzil, G.; Porst, M.; Schwind, K.H.; Dietsch, P.; Hilgers, K.F. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H203–H208. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A. Correlation between [Na+], [glycosaminoglycan] and Na+/K+ pump density in the extracellular matrix of bovine articular cartilage. Physiol. Res. 1998, 47, 47–52. [Google Scholar] [PubMed]
- Fischereder, M.; Michalke, B.; Schmöckel, E.; Habicht, A.; Kunisch, R.; Pavelic, I.; Szabados, B.; Schönermarck, U.; Nelson, P.J.; Stangl, M. Sodium storage in human tissues is mediated by glycosaminoglycan expression. Am. J. Physiol. Renal Physiol. 2017, 313, F319–F325. [Google Scholar] [CrossRef] [PubMed]
- Thowsen, I.M.; Karlsen, T.V.; Nikpey, E.; Haslene-Hox, H.; Skogstrand, T.; Randolph, G.J.; Zinselmeyer, B.H.; Tenstad, O.; Wiig, H. Na (+) is shifted from the extracellular to the intracellular compartment and is not inactivated by glycosaminoglycans during high salt conditions in rats. J. Physiol. 2022, 600, 2293–2309. [Google Scholar] [CrossRef] [PubMed]
- Wenstedt, E.F.E.; Olde Engberink, R.H.G.; Vogt, L. Sodium Handling by the Blood Vessel Wall: Critical for Hypertension Development. Hypertension 2018, 71, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Olde Engberink, R.H.; Rorije, N.M.; Homan van der Heide, J.J.; van den Born, B.J.; Vogt, L. Role of the vascular wall in sodium homeostasis and salt sensitivity. J. Am. Soc. Nephrol. 2015, 26, 777–783. [Google Scholar] [CrossRef]
- Oberleithner, H.; Riethmüller, C.; Schillers, H.; MacGregor, G.A.; de Wardener, H.E.; Hausberg, M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc. Natl. Acad. Sci. USA 2007, 104, 16281–16286. [Google Scholar] [CrossRef]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflugers Arch. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Vlahu, C.A.; Lemkes, B.A.; Struijk, D.G.; Koopman, M.G.; Krediet, R.T.; Vink, H. Damage of the endothelial glycocalyx in dialysis patients. J. Am. Soc. Nephrol. 2012, 23, 1900–1908. [Google Scholar] [CrossRef]
- Vlahu, C.A.; Lopes Barreto, D.; Struijk, D.G.; Vink, H.; Krediet, R.T. Is the systemic microvascular endothelial glycocalyx in peritoneal dialysis patients related to peritoneal transport? Nephron. Clin. Pract. 2014, 128, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Hatayama, N.; Kamiya, K.; Yamauchi, M.; Kinashi, H.; Yamaguchi, M.; Katsuno, T.; Nobata, H.; Watanabe, K.; Wakatsuki, A.; et al. Roles of glomerular endothelial hyaluronan in the development of proteinuria. Physiol. Rep. 2021, 9, e15019. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Voumvourakis, A.; Makavos, G.; Triantafyllidi, H.; Pavlidis, G.; Katogiannis, K.; Benas, D.; Vlastos, D.; Trivilou, P.; Varoudi, M.; et al. Association of impaired endothelial glycocalyx with arterial stiffness, coronary microcirculatory dysfunction, and abnormal myocardial deformation in untreated hypertensives. J. Clin. Hypertens. 2018, 20, 672–679. [Google Scholar] [CrossRef]
- Nijst, P.; Verbrugge, F.H.; Grieten, L.; Dupont, M.; Steels, P.; Tang, W.H.W.; Mullens, W. The pathophysiological role of interstitial sodium in heart failure. J. Am. Coll. Cardiol. 2015, 65, 378–388. [Google Scholar] [CrossRef]
- Sugiyama, N.; Tawada, M.; Sun, T.; Suzuki, Y.; Kinashi, H.; Yamaguchi, M.; Katsuno, T.; Aten, J.; Vlahu, C.A.; van Kuppevelt, T.H.; et al. Low-GDP, pH-neutral solutions preserve peritoneal endothelial glycocalyx during long-term peritoneal dialysis. Clin. Exp. Nephrol. 2021, 25, 1035–1046. [Google Scholar] [CrossRef]
- Dong, J.; Li, Y.; Yang, Z.; Luo, J. Low dietary sodium intake increases the death risk in peritoneal dialysis. Clin. J. Am. Soc. Nephrol. 2010, 5, 240–247. [Google Scholar] [CrossRef]
- Ikenoue, T.; Koike, K.; Fukuma, S.; Ogata, S.; Iseki, K.; Fukuhara, S. Salt Intake and All-Cause Mortality in Hemodialysis Patients. Am. J. Nephrol. 2018, 48, 87–95. [Google Scholar] [CrossRef]
- Suzuki, N.; Hitomi, Y.; Takata, H.; Ushiya, S.; Yamada, M.; Sakai, Y.; Konishi, T.; Takeda, Y.; Sumino, Y.; Mizo, M.; et al. Association between salt intake and long-term mortality in hemodialysis patients: A retrospective cohort study. PLoS ONE 2021, 16, e0260671. [Google Scholar] [CrossRef]
- D’Elia, L.; Manfredi, M.; Strazzullo, P.; Galletti, F.; MINISAL-SIIA Study Group. Validation of an easy questionnaire on the assessment of salt habit: The MINISAL-SIIA Study Program. Eur. J. Clin. Nutr. 2019, 73, 793–800. [Google Scholar] [CrossRef]
- Morris, R.C.; Jr Sebastian, A.; Forman, A.; Tanaka, M.; Schmidlin, O. Normotensive salt sensitivity: Effects of race and dietary potassium. Hypertension 1999, 33, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Iseki, K.; Uehara, H.; Nishime, K.; Tokuyama, K.; Yoshihara, K.; Kinjo, K.; Shiohira, Y.; Fukiyama, K. Impact of the initial levels of laboratory variables on survival in chronic dialysis patients. Am. J. Kidney Dis. 1996, 28, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Rhee, C.M.; Streja, E.; Soohoo, M.; Obi, Y.; Chou, J.A.; Tortorici, A.R.; Ravel, V.A.; Kovesdy, C.P.; Kalantar-Zadeh, K. Racial and Ethnic Differences in Mortality Associated with Serum Potassium in a Large Hemodialysis Cohort. Am. J. Nephrol. 2017, 45, 509–521. [Google Scholar] [CrossRef]
- Narasaki, Y.; Okuda, Y.; Kalantar, S.S.; You, A.S.; Novoa, A.; Nguyen, T.; Streja, E.; Nakata, T.; Colman, S.; Kalantar-Zadeh, K.; et al. Dietary Potassium Intake and Mortality in a Prospective Hemodialysis Cohort. J. Ren. Nutr. 2021, 31, 411–420. [Google Scholar] [CrossRef] [PubMed]
- St-Jules, D.E.; Goldfarb, D.S.; Sevick, M.A. Nutrient Non-equivalence: Does Restricting High-Potassium Plant Foods Help to Prevent Hyperkalemia in Hemodialysis Patients? J. Ren. Nutr. 2016, 26, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Bernier-Jean, A.; Wong, G.; Saglimbene, V.; Ruospo, M.; Palmer, S.C.; Natale, P.; Garcia-Larsen, V.; Johnson, D.W.; Tonelli, M.; Hegbrant, J.; et al. Dietary Potassium Intake and All-Cause Mortality in Adults Treated with Hemodialysis. Clin. J. Am. Soc. Nephrol. 2021, 16, 1851–1861. [Google Scholar] [CrossRef]
- Adrogué, H.J.; Madias, N.E. Sodium and potassium in the pathogenesis of hypertension. N. Engl. J. Med. 2007, 356, 1966–1978. [Google Scholar] [CrossRef]
- Hammon, M.; Grossmann, S.; Linz, P.; Kopp, C.; Dahlmann, A.; Garlichs, C.; Janka, R.; Cavallaro, A.; Luft, F.C.; Uder, M.; et al. 23Na Magnetic Resonance Imaging of the Lower Leg of Acute Heart Failure Patients during Diuretic Treatment. PLoS ONE 2015, 10, e0141336. [Google Scholar] [CrossRef]
- Karg, M.V.; Bosch, A.; Kannenkeril, D.; Striepe, K.; Ott, C.; Schneider, M.P.; Boemke-Zelch, F.; Linz, P.; Nagel, A.M.; Titze, J.; et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: A randomised controlled trial. Cardiovasc. Diabetol. 2018, 17, 5. [Google Scholar] [CrossRef]
- Pergola, P.E.; Devalaraja, M.; Fishbane, S.; Chonchol, M.; Mathur, V.S.; Smith, M.T.; Lo, L.; Herzog, K.; Kakkar, R.; Davidson, M.H.; et al. Ziltivekimab for Treatment of Anemia of Inflammation in Patients on Hemodialysis: Results from a Phase 1/2 Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Soc. Nephrol. 2021, 32, 211–222. [Google Scholar] [CrossRef]
- Hung, A.M.; Ellis, C.D.; Shintani, A.; Booker, C.; Ikizler, T.A. IL-1β receptor antagonist reduces inflammation in hemodialysis patients. J. Am. Soc. Nephrol. 2011, 22, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.M.; Tsuchida, Y.; Nowak, K.L.; Sarkar, S.; Chonchol, M.; Whitfield, V.; Salas, N.; Dikalova, A.; Yancey, P.G.; Huang, J.; et al. IL-1 Inhibition and Function of the HDL-Containing Fraction of Plasma in Patients with Stages 3 to 5 CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Chonchol, M.; Ikizler, T.A.; Farmer-Bailey, H.; Salas, N.; Chaudhry, R.; Wang, W.; Smits, G.; Tengesdal, I.; Dinarello, C.A.; et al. IL-1 Inhibition and Vascular Function in CKD. J. Am. Soc. Nephrol. 2017, 28, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Kasai, K.; Imai, H. Novel low Na peritoneal dialysis solutions designed to optimize Na gap of effluent: Kinetics of Na and water removal. Perit. Dial. Int. 2009, 29, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, S.; Salerno, F.R.; Akbari, A.; McIntyre, C.W. Influence of Dialysate Sodium Prescription on Skin and Muscle Sodium Concentration. Am. J. Kidney Dis. 2021, 78, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.M.; Limkunakul, C.; Placido, J.S.; Siew, E.D.; Ellis, C.D.; Shintani, A.; Ikizler, T.A. Administration of IL-1ra improves adiponectin levels in chronic hemodialysis patients. J. Nephrol. 2014, 27, 681–688. [Google Scholar] [CrossRef]
- Abdelmageed, M.M.; Kefaloyianni, E.; Arthanarisami, A.; Komaru, Y.; Atkinson, J.J.; Herrlich, A. TNF or EGFR inhibition equally block AKI-to-CKD transition: Opportunities for etanercept treatment. Nephrol. Dial. Transplant. 2023, 38, 1139–1150. [Google Scholar] [CrossRef]



| Year | Study Design | Subjects | Findings | Refs |
|---|---|---|---|---|
| 2012 | Cross-sectional | Normotensive subjects (17 men, 13 women) and 5 patients with primary aldosteronism | 29% increase in muscle sodium content in patients with aldosteronism compared with normal subjects. | [30] |
| 2013 | Cross-sectional | 56 healthy control men and women, 57 men and women with essential hypertension | Age-dependent increases in sodium content was observed in calf muscle in men. Patients with refractory hypertension showed increased tissue sodium content compared with normotensive controls. | [31] |
| 2015 | Cross-sectional | 24 HD patients and 27 age-matched healthy controls, 20 HD patients before and shortly after HD | Age was associated with higher tissue sodium content in controls. Older HD patients showed increased sodium and water in skin and muscle compared with age-matched controls. After HD, patients with low circulating VEGF-C levels showed higher skin sodium content compared with HD patients with high VEGF-C levels. | [32] |
| 2017 | Cross-sectional | 99 patients with mild to moderate CKD (42 women) | Skin sodium content, but not total overhydration, correlated with systolic blood pressure. Skin sodium content was closely linked to left ventricular mass in CKD patients. | [33] |
| 2019 | Cross-sectional | 23 patients with CKD5, 11 healthy controls | CKD patients had fluid overload when compared to controls. Tissue sodium concentrations were higher in the subcutaneous compartment, but not in the muscle. Tissue sodium content was correlated with fluid overload. Fluid overload in CKD was linked to higher IL-8 and inversely associated with E-selectin. Higher subcutaneous sodium concentration was linked to higher ICAM. | [34] |
| 2020 | Cross-sectional | 10 healthy controls, 20 patients with CKD5 (not on dialysis) | CKD patients had higher sodium and lower extracellular resistance. Tissue sodium concentration has an inverse linear relationship with extracellular resistance. | [35] |
| 2020 | Cross-sectional | 10 healthy controls, 12 CKD3-5, 13 HD, 10 PD patients | Tissue sodium in the skin, sleus, and tibia was higher in HD and PD patients compared with controls. Serum albumin showed a negative correlation with soleus sodium in HD patients. Estimated GFR showed a negative correlation with tissue sodium in merged control-CKD patients. Hemoglobin was negatively correlated with tissue sodium concentration in CKD and HD patients. | [36] |
| 2021 | Cross-sectional | 162 subjects (10 PD, 33 HD patients, 119 controls) | Patients on PD and HD showed higher muscle and skin sodium accumulation compared with controls. African American patients, older age, and male sex were associated with increased sodium content. Greater ultrafiltration was associated with lower skin sodium content in PD patients. Higher plasma IL-6 and hsCRP levels correlated with increased muscle and skin sodium content in the subjects. | [37] |
| 2021 | Prospective | 18 patients with HF, 34 HD patients, 31 patients with CKD (GFR matched to the HF patients) | HF patients showed higher skin sodium content than matched CKD patients, which was indistinguishable from skin sodium content in HD patients. | [38] |
| 2022 | Cross-sectional | 52 HD patients divided into groups with (23 subjects) or without (29 subjects) a positive history of cardiovascular events | HD patients with previous CVD events showed an increased sodium content in skin and muscle tissue compared to HD patients without CVD events. Fluid amount was not different between groups. Tissue sodium accumulation in HD-CVD patients was paralleled by higher plasma IL-6 levels. | [39] |
| 2022 | Cross-sectional | 36 pediatric participants (17 healthy, 19 CKD) and 19 healthy adults | Healthy adults had higher tissue sodium content compared with pediatric groups. No differences in tissue sodium content were found between healthy children and CKD patients. CKD patients with glomerular disease showed increased sodium content; however, CKD patients with tubular disorders showed reduced sodium content. | [40] |
| 2022 | Prospective | 52 subjects (10 PD, 42 HD patients) | Higher skin sodium accumulation was associated with worse clinical outcomes. Skin sodium concentration could be a predictor of survival. | [41] |
| Year | Model | Findings | Refs |
|---|---|---|---|
| 2011 | Subtotal nephrectomized rats treated with NaCl drinking water | NaCl-loaded subtotal nephrectomy caused LVH, hypertension, and increased plasma levels of PTH, creatinine, inorganic phosphorus, ADMA, and IL-6. | [71] |
| 2013 | 5/6 nephrectomized rats with nitric oxide depletion and a high-salt diet | Renal failure developed by 12 weeks after subtotal nephrectomy. Inflammation-related tubulo-interstitial damage and fibrosis, tubular atrophy, and focal glomerulosclerosis led to massive reduction of healthy glomeruli within the remnant kidney. | [72] |
| 2015 | 5/6 nephrectomized rats with high-salt diet | High salt activated the intrarenal and cerebral renin-angiotensin axes, which is related to the promotion of oxidative stress, renal fibrosis, and progression of CKD. | [73] |
| 2017 | 5/6 nephrectomized mice with salt loading | Salt loading induced macrophage infiltration in the peritoneal wall, heart, and para-aortic tissues through the TonEBP–MCP-1 pathway. | [50] |
| 2019 | 5/6 nephrectomized mice with salt loading | Salt loading induced peritoneal inflammation, angiogenesis, and high peritoneal transport rate. Blockade of IL-6 signaling rescued peritoneal transport function. | [74] |
| 2020 | Rats treated with nitric oxide synthase inhibition and a high-salt diet | Early treatment with NF-κB inhibitor prevented the development of late renal injury and inflammation. | [75] |
| 2021 | High-salt-load 5/6 nephrectomized rats | Inhibition of central PRR expression reduced inflammation and oxidative stress in both brain and kidney and ameliorated renal injury and fibrosis. The central MAPK/ERK1/2 and PI3K/Akt signaling pathways were related to the mechanism as well as the angiotensin-converting enzyme 1–angiotensin II–angiotensin type 1 receptors axis. | [76] |
| 2021 | 5/6 nephrectomized rats with NaCl treatment | NaCl treatment induced a pro-inflammatory phenotype in peritoneal macrophages derived from nephrectomized rats. High salt intake promoted immune activation of macrophages through phosphorylation of STAT1. | [77] |
| 2022 | 5/6 nephrectomized mice with salt loading | Salt loading induced cardiac inflammation and fibrosis along with elevated levels of proinflammatory cytokines. Blockade of IL-6 signaling showed anti-inflammatory, antifibrotic, and partial antioxidative effects on the heart. | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, Y.; Sun, T.; Tanaka, H.; Yamaguchi, M.; Kinashi, H.; Sakata, F.; Kunoki, S.; Sakai, Y.; Ishimoto, T. Tissue Sodium Accumulation Induces Organ Inflammation and Injury in Chronic Kidney Disease. Int. J. Mol. Sci. 2023, 24, 8329. https://doi.org/10.3390/ijms24098329
Ito Y, Sun T, Tanaka H, Yamaguchi M, Kinashi H, Sakata F, Kunoki S, Sakai Y, Ishimoto T. Tissue Sodium Accumulation Induces Organ Inflammation and Injury in Chronic Kidney Disease. International Journal of Molecular Sciences. 2023; 24(9):8329. https://doi.org/10.3390/ijms24098329
Chicago/Turabian StyleIto, Yasuhiko, Ting Sun, Hiroya Tanaka, Makoto Yamaguchi, Hiroshi Kinashi, Fumiko Sakata, Shunnosuke Kunoki, Yukinao Sakai, and Takuji Ishimoto. 2023. "Tissue Sodium Accumulation Induces Organ Inflammation and Injury in Chronic Kidney Disease" International Journal of Molecular Sciences 24, no. 9: 8329. https://doi.org/10.3390/ijms24098329