
The Cytokine CX3CL1 and ADAMs/MMPs in Concerted Cross-Talk Influencing Neurodegenerative Diseases

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
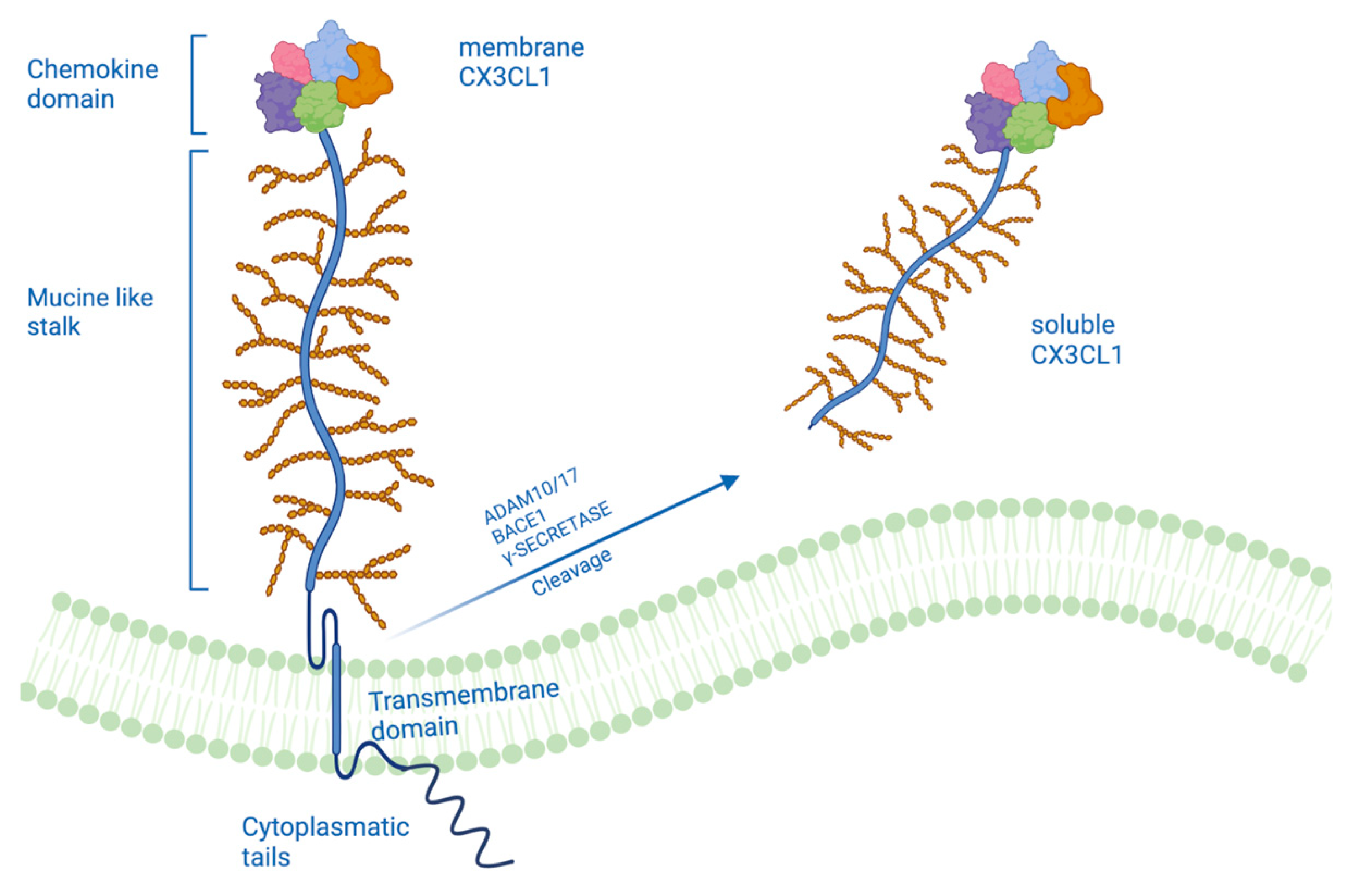
2. A Different Form of CX3CL1
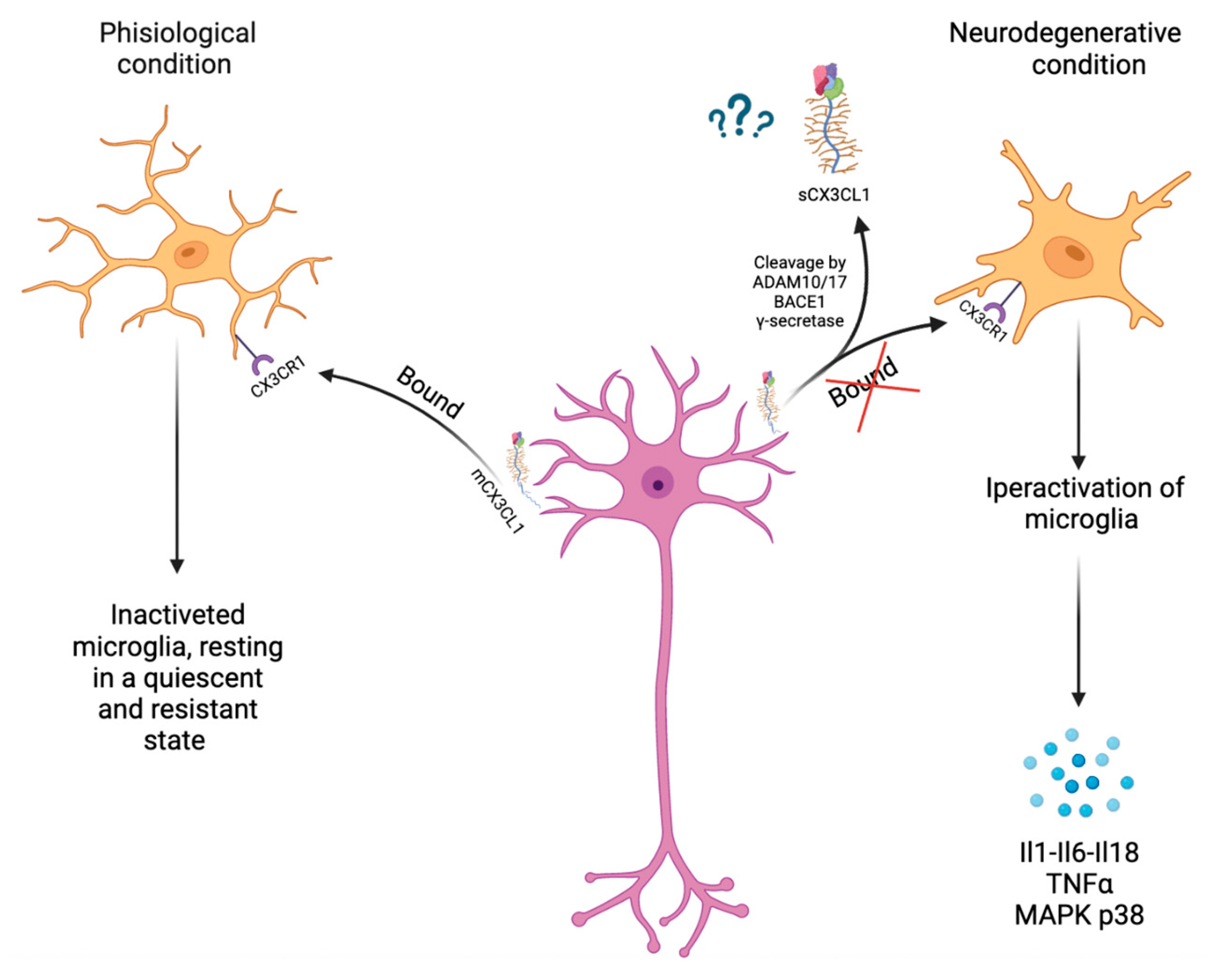
3. CX3CL1 and ADAMs/MMPs Involved in Neuroinflammation
4. CX3CL1 and ADAMs/MMPs in Neurodegenerative Disease
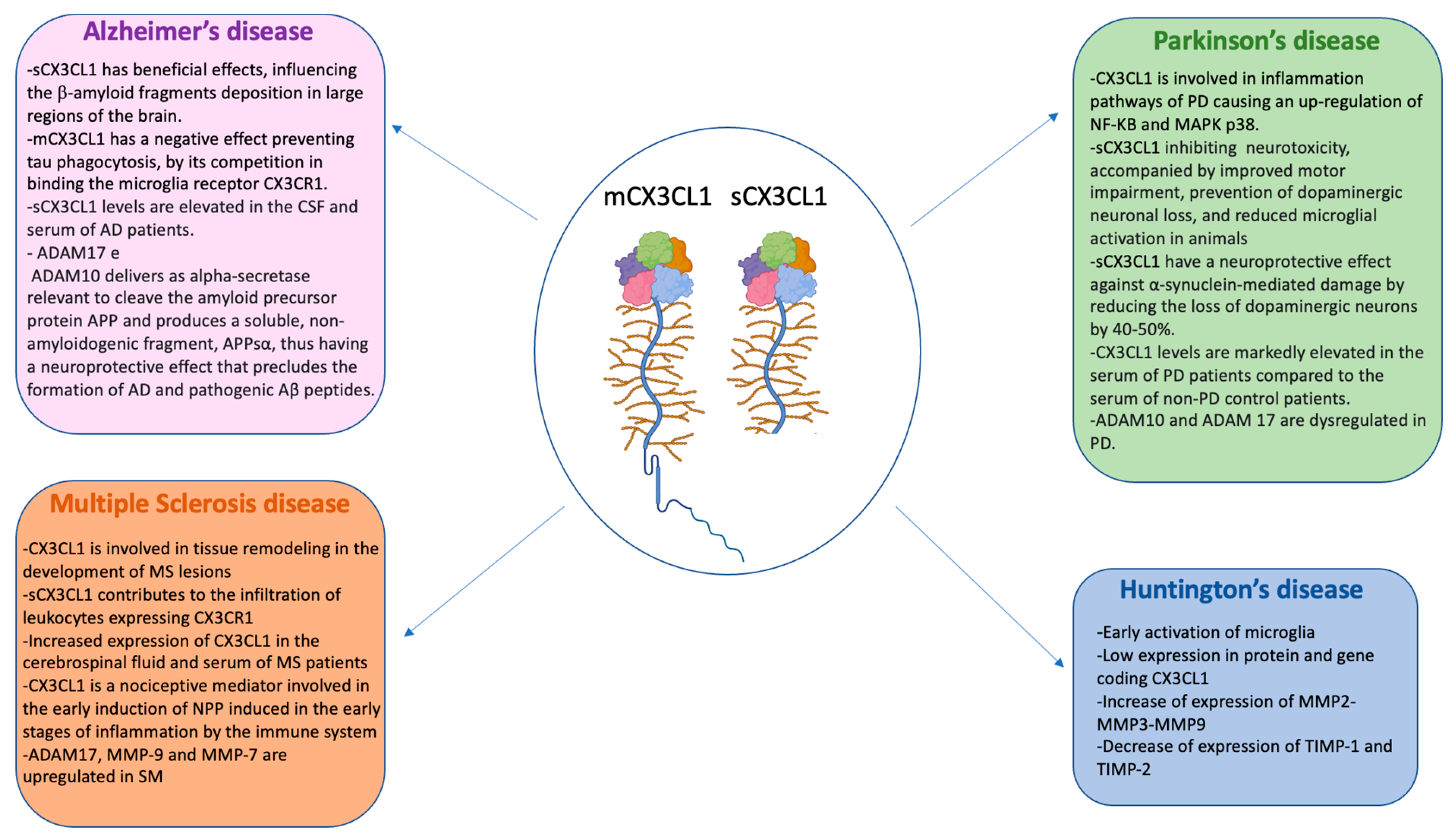
4.1. CX3CL1 and ADAMs/MMPs in Alzheimer’s Disease
4.2. CX3CL1 and ADAMs/MMPs in Parkinson’s Disease
4.3. CX3CL1 and ADAMs/MMPs in Multiple Sclerosis (SM)
4.4. CX3CL1 and MMPs in Huntington’s Disease
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A Potential New Target for Inflammatory Diseases. Mol. Interv. 2010, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Luster, A.D. Chemokines—Chemotactic Cytokines That Mediate Inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, M.; Zhao, X. Role of chemokine systems in cancer and inflammatory diseases. Medcomm 2022, 3, e147. [Google Scholar] [CrossRef]
- Liu, W.; Bian, C.; Liang, Y.; Jiang, L.; Qian, C.; Dong, J. CX3CL1: A potential chemokine widely involved in the process spinal metastases. Oncotarget 2017, 8, 15213–15219. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef]
- Umehara, H.; Bloom, E.T.; Okazaki, T.; Nagano, Y.; Yoshie, O.; Imai, T. Fractalkine in Vascular Biology: From basic research to clinical disease. Arter. Thromb. Vasc. Biol. 2004, 24, 34–40. [Google Scholar] [CrossRef]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef]
- O’sullivan, S.A.; Gasparini, F.; Mir, A.K.; Dev, K.K. Fractalkine shedding is mediated by p38 and the ADAM10 protease under pro-inflammatory conditions in human astrocytes. J. Neuroinflamm. 2016, 13, 189. [Google Scholar] [CrossRef]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D.; et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 2016, 5, e12748. [Google Scholar] [CrossRef]
- Costa, S.; Ragusa, M.A.; Buglio, G.L.; Scilabra, S.D.; Nicosia, A. The Repertoire of Tissue Inhibitors of Metalloproteases: Evolution, Regulation of Extracellular Matrix Proteolysis, Engineering and Therapeutic Challenges. Life 2022, 12, 1145. [Google Scholar] [CrossRef]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648.e7. [Google Scholar] [CrossRef]
- Fan, Q.; Gayen, M.; Singh, N.; Gao, F.; He, W.; Hu, X.; Tsai, L.-H.; Yan, R. The intracellular domain of CX3CL1 regulates adult neurogenesis and Alzheimer’s amyloid pathology. J. Exp. Med. 2019, 216, 1891–1903. [Google Scholar] [CrossRef]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor Necrosis Factor-α-converting Enzyme (ADAM17) Mediates the Cleavage and Shedding of Fractalkine (CX3CL1)*. J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Nakamura, M.; Satoh, H.; Saitoh, H.; Segawa, I.; Hiramori, K. Expression of tumor necrosis factor-alpha–converting enzyme and tumor necrosis factor-alpha in human myocarditis. J. Am. Coll. Cardiol. 2000, 36, 1288–1294. [Google Scholar] [CrossRef]
- Patel, I.R.; Attur, M.G.; Patel, R.N.; Stuchin, S.A.; Abagyan, R.A.; Abramson, S.B.; Amin, A.R. TNF-α Convertase Enzyme from Human Arthritis-Affected Cartilage: Isolation of cDNA by Differential Display, Expression of the Active Enzyme, and Regulation of TNF-α. J. Immunol. 1998, 160, 4570–4579. [Google Scholar] [CrossRef] [PubMed]
- Schulte, A.; Schulz, B.; Andrzejewski, M.; Hundhausen, C.; Mletzko, S.; Achilles, J.; Reiss, K.; Paliga, K.; Weber, C.; John, S.R.; et al. Sequential processing of the transmembrane chemokines CX3CL1 and CXCL16 by α- and γ-secretases. Biochem. Biophys. Res. Commun. 2007, 358, 233–240. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Manuel, M.N.; Mi, D.; Mason, J.O.; Price, D.J. Regulation of cerebral cortical neurogenesis by the Pax6 transcription factor. Front. Cell. Neurosci. 2015, 9, 70. [Google Scholar] [CrossRef]
- Lu, J.; Wu, Y.; Sousa, N.; Almeida, O.F.X. SMAD pathway mediation of BDNF and TGFβ2 regulation of proliferation and differentiation of hippocampal granule neurons. Development 2005, 132, 3231–3242. [Google Scholar] [CrossRef]
- Massagué, J.; Xi, Q. TGF-β control of stem cell differentiation genes. FEBS Lett. 2012, 586, 1953–1958. [Google Scholar] [CrossRef]
- Winter, A.N.; Subbarayan, M.S.; Grimmig, B.; Weesner, J.A.; Moss, L.; Peters, M.; Weeber, E.; Nash, K.; Bickford, P.C. Two forms of CX3CL1 display differential activity and rescue cognitive deficits in CX3CL1 knockout mice. J. Neuroinflamm. 2020, 17, 157. [Google Scholar] [CrossRef]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.-W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef]
- Biber, K.; Vinet, J.; Boddeke, H. Neuron-microglia signaling: Chemokines as versatile messengers. J. Neuroimmunol. 2008, 198, 69–74. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Maciejewski-Lenoir, D.; Chen, S.; Feng, L.; Maki, R.; Bacon, K.B. Related Content Characterization of Fractalkine in Rat Brain Cells: Migratory and Activation Signals for CX 3 CR-1-Expressing Microglia Combinatorial Model of Chemokine Involvement in Glomerular Monocyte Recruitment: Role of CXC Chemokine Receptor 2 in Innltration During Nephrotoxic Nephritis. J. Immunol. 1999, 163, 1628–1635. [Google Scholar] [CrossRef]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.-Y. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, e5. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Shen, X.; Wang, H. The Distinct Role of ADAM17 in APP Proteolysis and Microglial Activation Related to Alzheimer’s Disease. Cell. Mol. Neurobiol. 2015, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Hsia, H.-E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ’A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef] [PubMed]
- Subbarayan, M.S.; Joly-Amado, A.; Bickford, P.C.; Nash, K.R. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacol. Ther. 2021, 231, 107989. [Google Scholar] [CrossRef] [PubMed]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The Soluble Isoform of CX3CL1 Is Necessary for Neuroprotection in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef]
- Nash, K.R.; Moran, P.; Finneran, D.J.; Hudson, C.; Robinson, J.; Morgan, D.; Bickford, P.C. Fractalkine Over Expression Suppresses α-Synuclein-mediated Neurodegeneration. Mol. Ther. 2015, 23, 17–23. [Google Scholar] [CrossRef]
- Jiang, S.; Bhaskar, K. Dynamics of the Complement, Cytokine, and Chemokine Systems in the Regulation of Synaptic Function and Dysfunction Relevant to Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1123–1135. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Perea, J.R.; Jurado-Arjona, J.; Rábano, A.; Hernández, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef]
- Guedes, J.R.; Lao, T.; Cardoso, A.L.; El Khoury, J. Roles of Microglial and Monocyte Chemokines and Their Receptors in Regulating Alzheimer’s Disease-Associated Amyloid-β and Tau Pathologies. Front. Neurol. 2018, 9, 549. [Google Scholar] [CrossRef]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2019, 30, 9–30. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, F.; Liu, H.; Wei, F. NF-kB mediated CX3CL1 activation in the dorsal root ganglion contributes to the maintenance of neuropathic pain induced in adult male Sprague Dawley rats. Acta Cir. Bras. 2018, 33, 619–628. [Google Scholar] [CrossRef]
- Hwang, C.J.; Choi, D.-Y.; Park, M.H.; Hong, J.T. NF-κB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2017, 18, 3–10. [Google Scholar] [CrossRef]
- Shi, Z.-M.; Han, Y.-W.; Han, X.-H.; Zhang, K.; Chang, Y.-N.; Hu, Z.-M.; Qi, H.-X.; Ting, C.; Zhen, Z.; Hong, W. Upstream regulators and downstream effectors of NF-κB in Alzheimer’s disease. J. Neurol. Sci. 2016, 366, 127–134. [Google Scholar] [CrossRef]
- Kulczyńska-Przybik, A.; Słowik, A.; Mroczko, P.; Borawski, B.; Groblewska, M.; Borawska, R.; Mroczko, B. Cerebrospinal Fluid and Blood CX3CL1 as a Potential Biomarker in Early Diagnosis and Prognosis of Dementia. Curr. Alzheimer Res. 2020, 17, 709–721. [Google Scholar] [CrossRef]
- Nash, K.R.; Lee, D.C.; Hunt, J.B.; Morganti, J.M.; Selenica, M.-L.; Moran, P.; Reid, P.; Brownlow, M.; Yang, C.G.-Y.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef]
- Vincent, B.; Cisse, M.; Sunyach, C.; Guillot-Sestier, M.-V.; Checler, F. Regulation of βAPP and PrPc Cleavage by α-Secretase: Mechanistic and Therapeutic Perspectives. Curr. Alzheimer Res. 2008, 5, 202–211. [Google Scholar] [CrossRef]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef]
- Hartl, D.; Aesg; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; et al. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry 2018, 25, 629–639. [Google Scholar] [CrossRef]
- Kim, M.; Suh, J.; Romano, D.; Truong, M.H.; Mullin, K.; Hooli, B.; Norton, D.; Tesco, G.; Elliott, K.; Wagner, S.L.; et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum. Mol. Genet. 2009, 18, 3987–3996. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Liu, K.-N.; Luo, Y.; Slack, J.L.; Stocking, K.L.; Peschon, J.J.; Johnson, R.S.; Castner, B.J.; Cerretti, D.P.; Black, R.A. Evidence That Tumor Necrosis Factor α Converting Enzyme Is Involved in Regulated α-Secretase Cleavage of the Alzheimer Amyloid Protein Precursor. J. Biol. Chem. 1998, 273, 27765–27767. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-S.; Yang, S.; Tan, Z.-X.; Wang, M.-M.; Xing, Y.; Dong, F.; Zhang, F. The benefits and mechanisms of exercise training for Parkinson’s disease. Life Sci. 2020, 245, 117345. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Anagnostouli, M.; Chrousos, G.P.; Bougea, A. Massage therapy as a complementary treatment for Parkinson’s disease: A Systematic Literature Review. Complement. Ther. Med. 2020, 49, 102340. [Google Scholar] [CrossRef]
- Pabon, M.M.; Bachstetter, A.D.; Hudson, C.E.; Gemma, C.; Bickford, P.C. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflamm. 2011, 8, 9. [Google Scholar] [CrossRef]
- Tristão, F.S.M.; Lazzarini, M.; Martin, S.; Amar, M.; Stühmer, W.; Kirchhoff, F.; Gomes, L.A.C.; Lanfumey, L.; Prediger, R.D.; Sepulveda, J.E.; et al. CX3CR1 Disruption Differentially Influences Dopaminergic Neuron Degeneration in Parkinsonian Mice Depending on the Neurotoxin and Route of Administration. Neurotox. Res. 2016, 29, 364–380. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Zhao, A.; Luo, N.; Niu, M.; Kang, W.; Xie, A.; Lu, H.; Chen, L.; Liu, J. Parkinson’s disease peripheral immune biomarker profile: A multicentre, cross-sectional and longitudinal study. J. Neuroinflamm. 2022, 19, 116. [Google Scholar] [CrossRef]
- Gupta, M.; Paliwal, V.K.; Babu, G.N. Serum fractalkine and 3-nitrotyrosine levels correlate with disease severity in Parkinson’s disease: A pilot study. Metab. Brain Dis. 2022, 37, 209–217. [Google Scholar] [CrossRef]
- Zhou, M.; Lin, Y.; Lu, L.; Zhang, Z.; Guo, W.; Peng, G.; Zhang, W.; Zhu, Z.; Wu, Z.; Mo, M.; et al. Association of ADAM10 gene variants with sporadic Parkinson’s disease in Chinese Han population. J. Gene Med. 2021, 23, e3319. [Google Scholar] [CrossRef]
- Li, W.W.; Shen, Y.Y.; Chen, D.W.; Li, H.Y.; Shi, Q.Q.; Mei, J.; Yang, H.; Zhou, F.; Shi, A.; Zhang, T.; et al. Genetic Association Between NGFR, ADAM17 Gene Polymorphism, and Parkinson’s Disease in the Chinese Han Population. Neurotox. Res. 2019, 36, 463–471. [Google Scholar] [CrossRef]
- Lange, J.; Lunde, K.A.; Sletten, C.; Møller, S.G.; Tysnes, O.-B.; Alves, G.; Larsen, J.P.; Maple-Grødem, J. Association of aBACE1Gene Polymorphism with Parkinson’s Disease in a Norwegian Population. Park. Dis. 2015, 2015, 973298. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Hulshof, S.; van Haastert, E.S.; Kuipers, H.F.; Elsen, P.J.V.D.; de Groot, C.J.; van der Valk, P.; Ravid, R.; Biber, K. CX3CL1 and CX3CR1 Expression in Human Brain Tissue: Noninflammatory Control versus Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2003, 62, 899–907. [Google Scholar] [CrossRef]
- Sunnemark, D.; Eltayeb, S.; Nilsson, M.; Wallström, E.; Lassmann, H.; Olsson, T.; Berg, A.-L.; Ericsson-Dahlstrand, A. CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: Kinetics and cellular origin. J. Neuroinflamm. 2005, 2, 17. [Google Scholar] [CrossRef]
- Blauth, K.; Zhang, X.; Chopra, M.; Rogan, S.; Markovic-Plese, S. The role of fractalkine (CX3CL1) in regulation of CD4+ cell migration to the central nervous system in patients with relapsing–remitting multiple sclerosis. Clin. Immunol. 2015, 157, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Broux, B.; Pannemans, K.; Zhang, X.; Markovic-Plese, S.; Broekmans, T.; Eijnde, B.O.; Van Wijmeersch, B.; Somers, V.; Geusens, P.; van der Pol, S.; et al. CX3CR1 drives cytotoxic CD4+CD28− T cells into the brain of multiple sclerosis patients. J. Autoimmun. 2012, 38, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.H.; Alabanza, L.M.; A Mahamed, D.; Bynoe, M.S. Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2012, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Ridderstad Wollberg, A.; Ericsson-Dahlstrand, A.; Juréus, A.; Ekerot, P.; Simon, S.; Nilsson, M.; Wiklund, S.-J.; Berg, A.-L.; Ferm, M.; Sunnemark, D.; et al. Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5409–5414. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef]
- Zhu, W.; Acosta, C.; MacNeil, B.; Cortes, C.; Intrater, H.; Gong, Y.; Namaka, M. Elevated Expression of Fractalkine (CX3CL1) and Fractalkine Receptor (CX3CR1) in the Dorsal Root Ganglia and Spinal Cord in Experimental Autoimmune Encephalomyelitis: Implications in Multiple Sclerosis-Induced Neuropathic Pain. BioMed. Res. Int. 2013, 2013, 14. [Google Scholar] [CrossRef]
- Plumb, J.; McQuaid, S.; Cross, A.K.; Surr, J.; Haddock, G.; Bunning, R.A.D.; Woodroofe, N. Upregulation of ADAM-17 expression in active lesions in multiple sclerosis. Mult. Scler. J. 2006, 12, 375–385. [Google Scholar] [CrossRef]
- Kieseier, B.C.; Pischel, H.; Neuen-Jacob, E.; Tourtellotte, W.W.; Hartung, H.-P. ADAM-10 and ADAM-17 in the inflamed human CNS. Glia 2003, 42, 398–405. [Google Scholar] [CrossRef]
- Muri, L.; Leppert, D.; Grandgirard, D.; Leib, S.L. MMPs and ADAMs in neurological infectious diseases and multiple sclerosis. Cell. Mol. Life Sci. 2019, 76, 3097–3116. [Google Scholar] [CrossRef]
- Yong, V.W.; Power, C.; Forsyth, P.; Edwards, D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001, 2, 502–511. [Google Scholar] [CrossRef]
- Lindberg, R.L.P.; De Groot, C.J.A.; Montagne, L.; Freitag, P.; Van Der Valk, P.; Kappos, L.; Leppert, D. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain 2001, 124, 1743–1753. [Google Scholar] [CrossRef]
- Snowden, J.S. The Neuropsychology of Huntington’s Disease. Arch. Clin. Neuropsychol. 2017, 32, 876–887. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Illarioshkin, S.N.; Klyushnikov, S.A.; Vigont, V.A.; Seliverstov, Y.A.; Kaznacheyeva, E.V. Molecular Pathogenesis in Huntington’s Disease. Biochemistry 2018, 83, 1030–1039. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Bonchev, D. Network analysis of human post-mortem microarrays reveals novel genes, microRNAs, and mechanistic scenarios of potential importance in fighting huntington’s disease. Comput. Struct. Biotechnol. J. 2016, 14, 117–130. [Google Scholar] [CrossRef]
- Yang, H.-M.; Yang, S.; Huang, S.-S.; Tang, B.-S.; Guo, J.-F. Microglial Activation in the Pathogenesis of Huntington’s Disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef]
- Kim, A.; García-García, E.; Straccia, M.; Comella-Bolla, A.; Miguez, A.; Masana, M.; Alberch, J.; Canals, J.M.; Rodríguez, M.J. Reduced Fractalkine Levels Lead to Striatal Synaptic Plasticity Deficits in Huntington’s Disease. Front. Cell. Neurosci. 2020, 14, 163. [Google Scholar] [CrossRef]
- Naphade, S.; Embusch, A.; Madushani, K.L.; Ring, K.L.; Ellerby, L.M. Altered Expression of Matrix Metalloproteinases and Their Endogenous Inhibitors in a Human Isogenic Stem Cell Model of Huntington’s Disease. Front. Neurosci. 2018, 11, 736. [Google Scholar] [CrossRef]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix Metalloproteinases Are Modifiers of Huntingtin Proteolysis and Toxicity in Huntington’s Disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef]
- Duran-Vilaregut, J.; del Valle, J.; Manich, G.; Camins, A.; Pallàs, M.; Vilaplana, J.; Pelegrí, C. Role of matrix metalloproteinase-9 (MMP-9) in striatal blood-brain barrier disruption in a 3-nitropropionic acid model of Huntington’s disease. Neuropathol. Appl. Neurobiol. 2011, 37, 525–537. [Google Scholar] [CrossRef]
- Connolly, C.; Magnusson-Lind, A.; Lu, G.; Wagner, P.; Southwell, A.; Hayden, M.; Björkqvist, M.; Leavitt, B. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience 2016, 325, 74–88. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iemmolo, M.; Ghersi, G.; Bivona, G. The Cytokine CX3CL1 and ADAMs/MMPs in Concerted Cross-Talk Influencing Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 8026. https://doi.org/10.3390/ijms24098026
Iemmolo M, Ghersi G, Bivona G. The Cytokine CX3CL1 and ADAMs/MMPs in Concerted Cross-Talk Influencing Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(9):8026. https://doi.org/10.3390/ijms24098026
Chicago/Turabian StyleIemmolo, Matilda, Giulio Ghersi, and Giulia Bivona. 2023. "The Cytokine CX3CL1 and ADAMs/MMPs in Concerted Cross-Talk Influencing Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 9: 8026. https://doi.org/10.3390/ijms24098026