
Differential Modulation of Dendritic Cell Biology by Endogenous and Exogenous Aryl Hydrocarbon Receptor Ligands
 ,
,
Abstract
:1. Introduction
2. Results
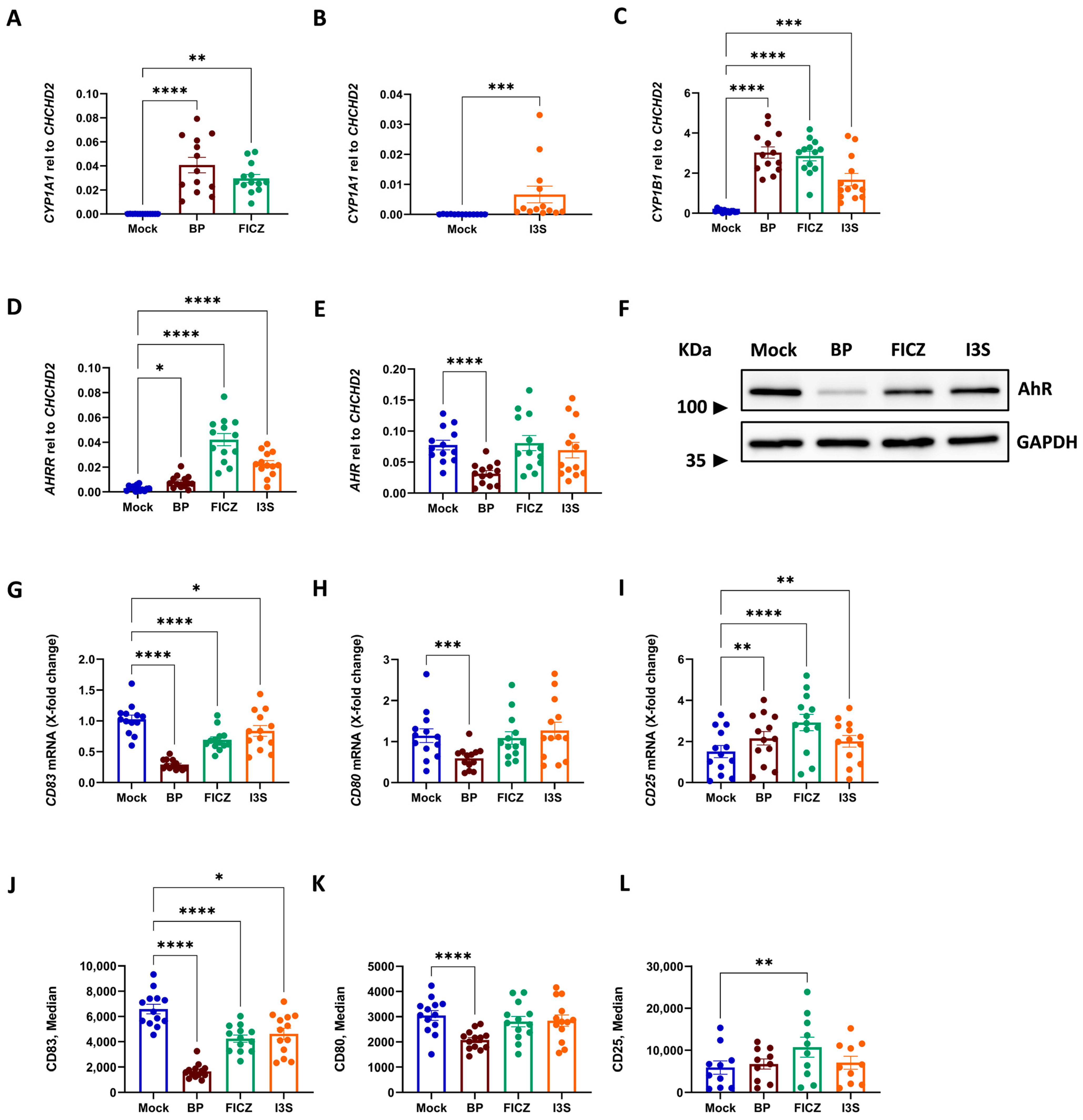
2.1. AhR Activation in DCs Results in Phenotypic Changes, Which Depend on the Class of AhR Ligand
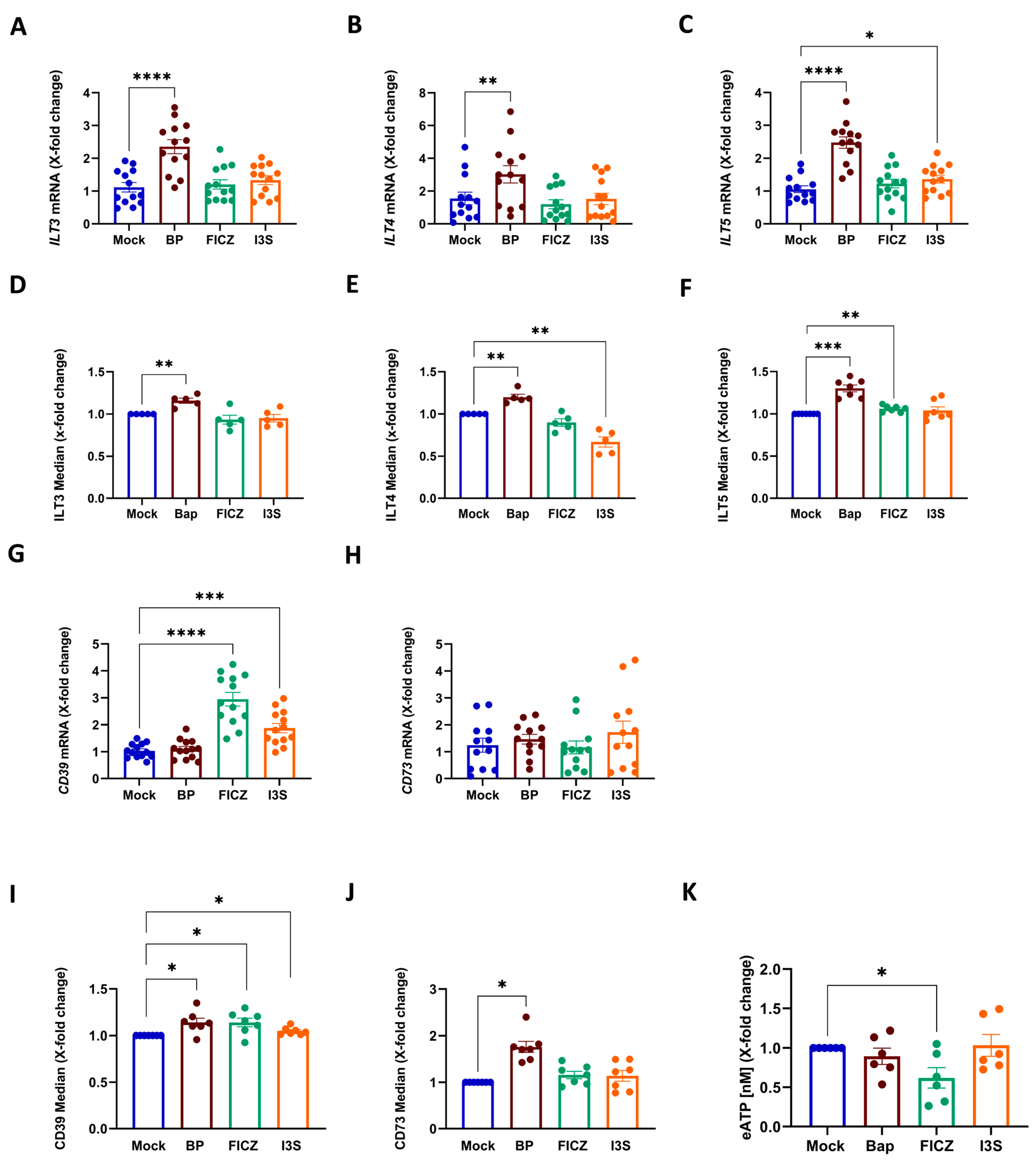
2.2. Induction of a Tolerogenic DC-Phenotype Is Ligand-Dependent
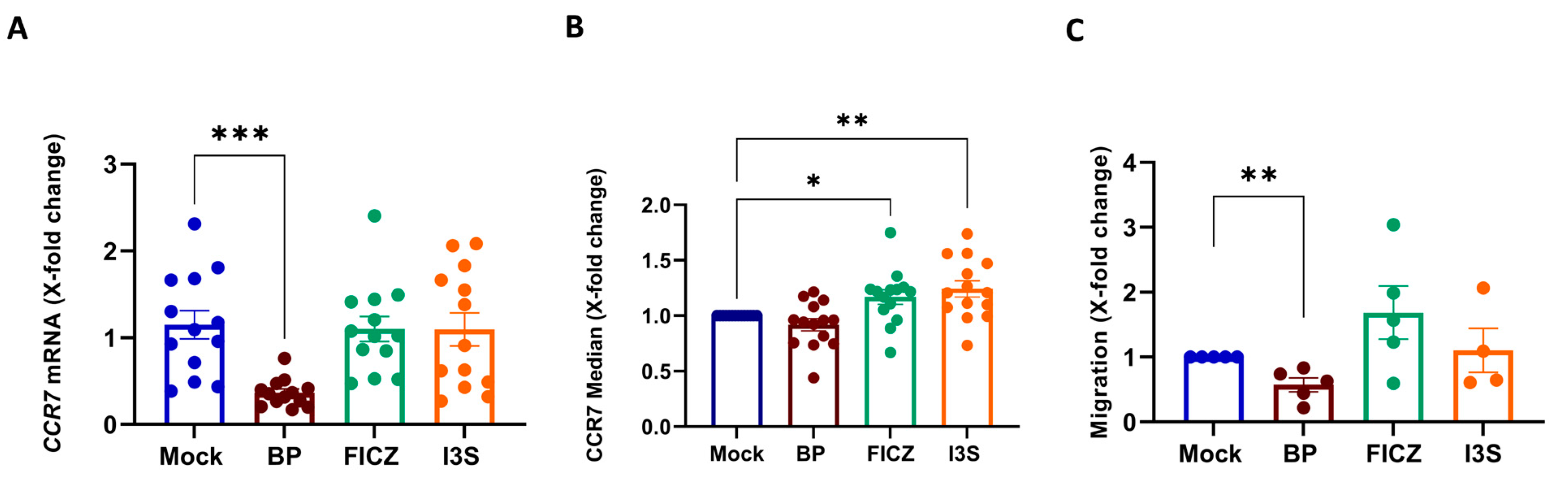
2.3. BP Treatment Impairs the Migratory Capacity of Mature Human DCs
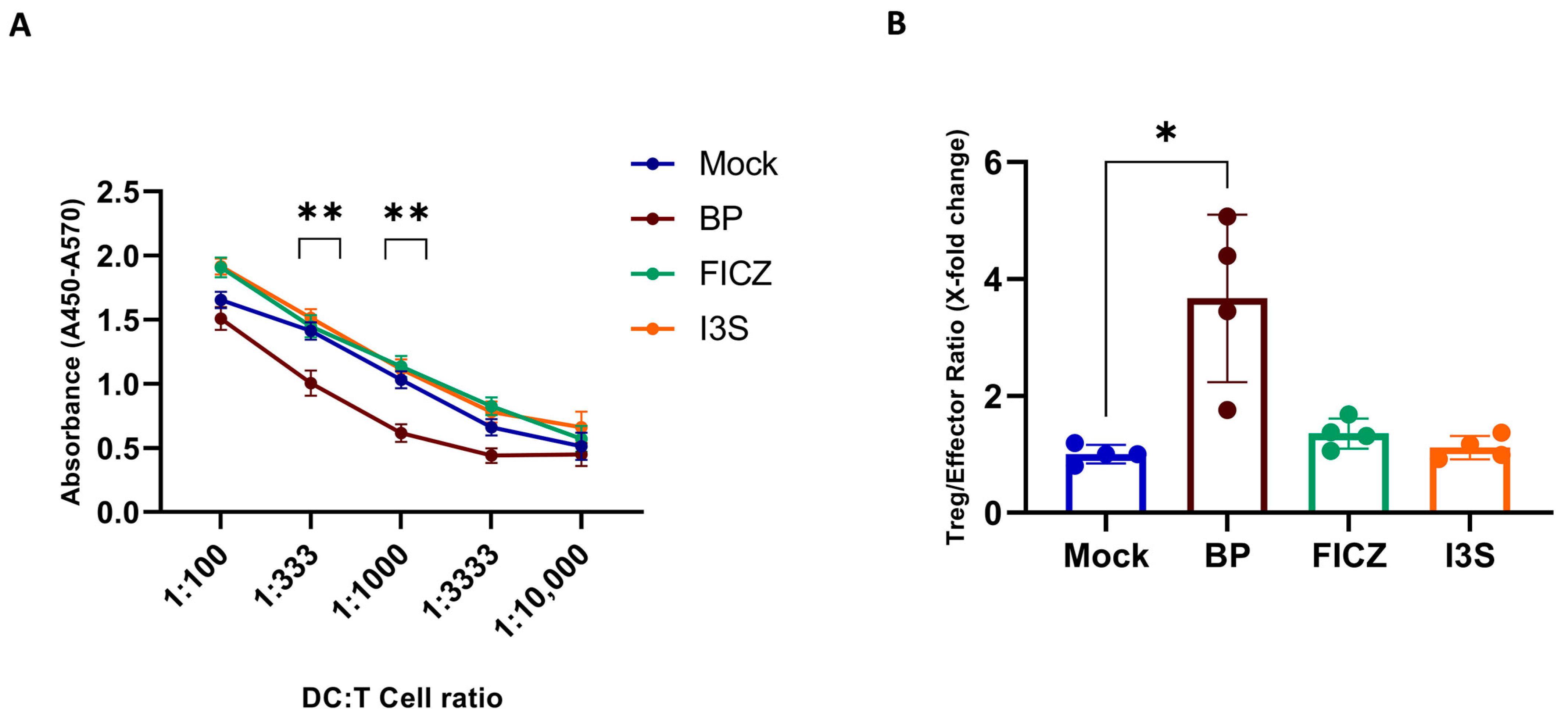
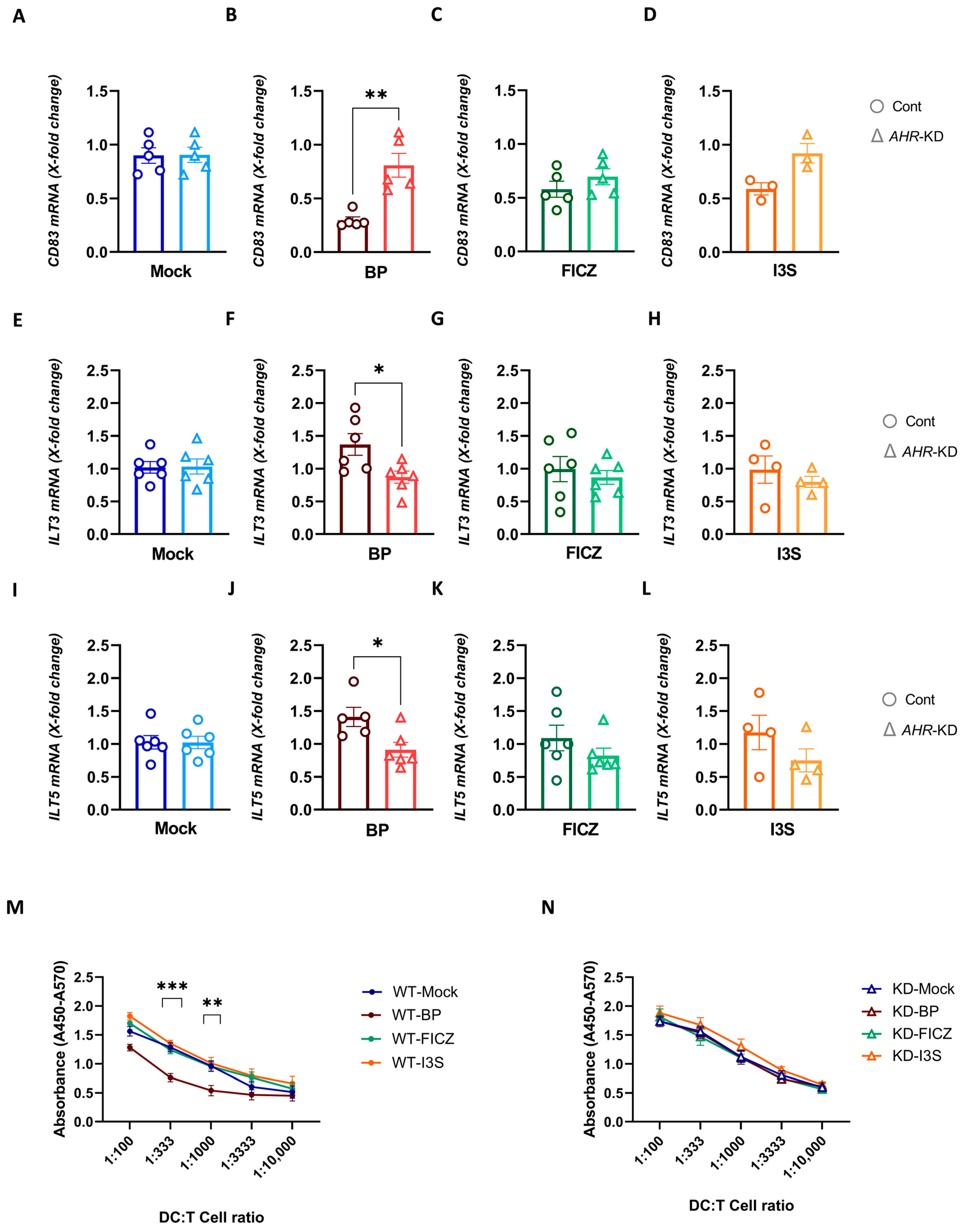
2.4. BP Treatment Induces a Regulatory DC Phenotype Which Correlates with Reduced T Cell Stimulatory Capacity
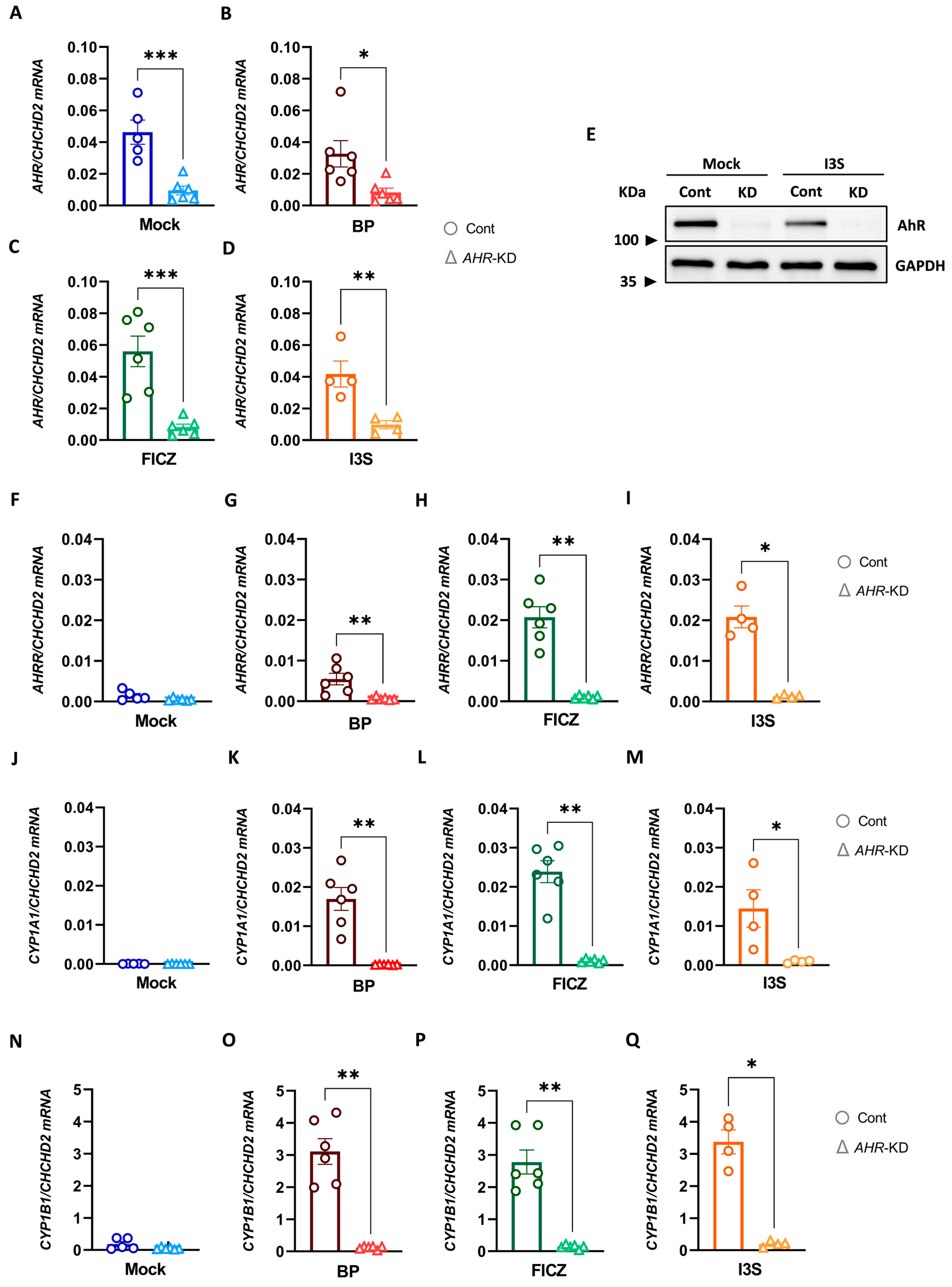
2.5. Efficient CRISPR/Cas9-Mediated AHR Knock-Down in Human DCs
2.6. Deletion of AHR in DCs Reverses the Tolerogenic Effects of BP
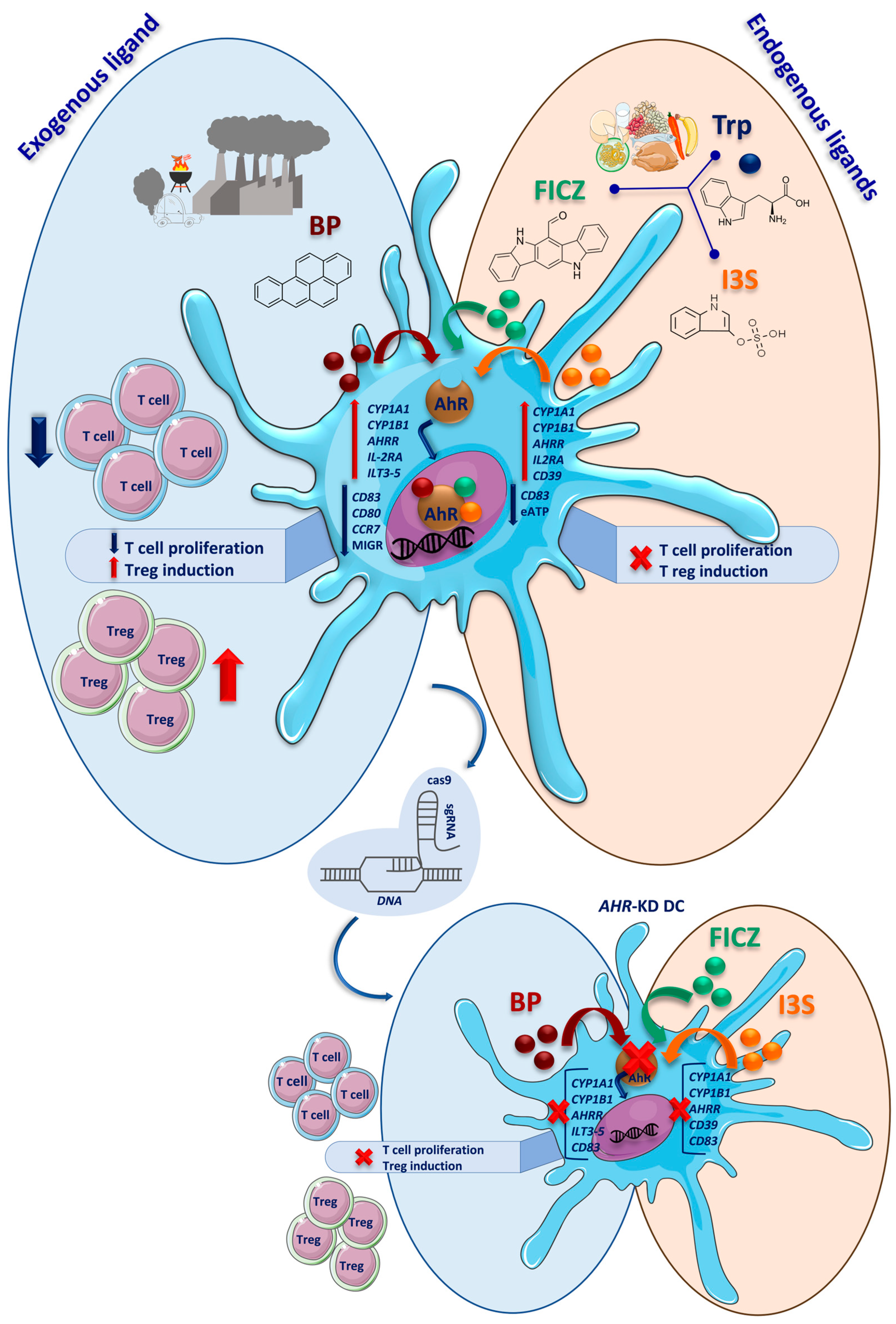
3. Discussion
4. Materials and Methods
4.1. Generation of Human Monocyte-Derived Dendritic Cells
4.2. Flow Cytometric Analysis
4.3. ATP Measurement
4.4. Transwell Migration Assay
4.5. sgRNA Sequences
4.6. CRISPR/Cas9-Mediated AHR Knock-Down
4.7. Sequencing of Edited DCs (Determination of Editing Efficiency)
4.8. Quantitative Real-Time PCR (qPCR)
4.9. Human Mixed Lymphocyte Reaction (MLR)
4.10. DC–T Cell Co-Cultures
4.11. Western Blot
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poland, A.; Glover, E.; Kende, A. Stereospecific, high affinity binding of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [CrossRef] [PubMed]
- Shinde, R.; McGaha, T.L. The aryl hydrocarbon receptor: Connecting immunity to the microenvironment. Trends Immunol. 2018, 39, 1005–1020. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Puccetti, P. The coevolution of IDO1 and AhR in the emergence of regulatory T-cells in mammals. Front. Immunol. 2015, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Durrin, L.K.; Jones, P.B.; Fisher, J.M.; Galeazzi, D.R.; Whitlock, J.P., Jr. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin receptors regulate transcription of the cytochrome P1-450 gene. J. Cell. Biochem. 1987, 35, 153–160. [Google Scholar] [CrossRef]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A molecular mechanism to switch the aryl hydrocarbon receptor from a transcription factor to an E3 ubiquitin ligase. Mol. Cell. Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef]
- Trikha, P.; Lee, D.A. The role of AhR in transcriptional regulation of immune cell development and function. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188335. [Google Scholar] [CrossRef]
- Baba, T.; Mimura, J.; Gradin, K.; Kuroiwa, A.; Watanabe, T.; Matsuda, Y.; Inazawa, J.; Sogawa, K.; Fujii-Kuriyama, Y. Structure and expression of the Ah receptor repressor gene. J. Biol. Chem. 2001, 276, 33101–33110. [Google Scholar] [CrossRef]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo [3, 2-b] carbazole is present in humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef]
- Effner, R.; Hiller, J.; Eyerich, S.; Traidl-Hoffmann, C.; Brockow, K.; Triggiani, M.; Behrendt, H.; Schmidt-Weber, C.B.; Buters, J. Cytochrome P450s in human immune cells regulate IL-22 and c-Kit via an AHR feedback loop. Sci. Rep. 2017, 7, srep44005. [Google Scholar] [CrossRef]
- Quintana, F.; Yeste, A.; Mascanfroni, I. Role and therapeutic value of dendritic cells in central nervous system autoimmunity. Cell Death Differ. 2015, 22, 215–224. [Google Scholar] [CrossRef]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef]
- Wild, A.B.; Krzyzak, L.; Peckert, K.; Stich, L.; Kuhnt, C.; Butterhof, A.; Seitz, C.; Mattner, J.; Grüner, N.; Gänsbauer, M. CD83 orchestrates immunity toward self and non-self in dendritic cells. JCI Insight 2019, 4, e126246. [Google Scholar] [CrossRef]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef]
- Michalski, J.; Deinzer, A.; Stich, L.; Zinser, E.; Steinkasserer, A.; Knippertz, I. Quercetin induces an immunoregulatory phenotype in maturing human dendritic cells. Immunobiology 2020, 225, 151929. [Google Scholar] [CrossRef]
- Juhasz, A.L.; Naidu, R. Bioremediation of high molecular weight polycyclic aromatic hydrocarbons: A review of the microbial degradation of benzo [a] pyrene. Int. Biodeterior. Biodegrad. 2000, 45, 57–88. [Google Scholar] [CrossRef]
- Verma, N.; Pink, M.; Rettenmeier, A.W.; Schmitz-Spanke, S. Review on proteomic analyses of benzo [a] pyrene toxicity. Proteomics 2012, 12, 1731–1755. [Google Scholar] [CrossRef]
- Wojdani, A.; Alfred, L.J. Alterations in cell-mediated immune functions induced in mouse splenic lymphocytes by polycyclic aromatic hydrocarbons. Cancer Res. 1984, 44, 942–945. [Google Scholar]
- Davila, D.R.; Romero, D.L.; Burchiel, S.W. Human T cells are highly sensitive to suppression of mitogenesis by polycyclic aromatic hydrocarbons and this effect is differentially reversed by α-naphthoflavone. Toxicol. Appl. Pharmacol. 1996, 139, 333–341. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Near, R.I.; Matulka, R.A.; Shneider, A.; Toselli, P.; Trombino, A.F.; Sherr, D.H. Activation of the aryl hydrocarbon receptor/transcription factor and bone marrow stromal cell-dependent preB cell apoptosis. J. Immunol. 1997, 158, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Laupeze, B.; Amiot, L.; Sparfel, L.; Le Ferrec, E.; Fauchet, R.; Fardel, O. Polycyclic aromatic hydrocarbons affect functional differentiation and maturation of human monocyte-derived dendritic cells. J. Immunol. 2002, 168, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-A.; Lee, J.-A.; Cheong, S.-W.; Youn, H.-J.; Park, J.-H. Benzo (a) pyrene inhibits growth and functional differentiation of mouse bone marrow-derived dendritic cells: Downregulation of RelB and eIF3 p170 by benzo (a) pyrene. Toxicol. Lett. 2007, 169, 82–90. [Google Scholar] [CrossRef]
- Rannug, A.; Rannug, U. The tryptophan derivative 6-formylindolo [3, 2-b] carbazole, FICZ, a dynamic mediator of endogenous aryl hydrocarbon receptor signaling, balances cell growth and differentiation. Crit. Rev. Toxicol. 2018, 48, 555–574. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.-C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Vogel, C.F.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2, 3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef]
- Pauly, S.K.; Fechner, J.H.; Zhang, X.; Torrealba, J.; Bradfield, C.A.; Mezrich, J.D. The aryl hydrocarbon receptor influences transplant outcomes in response to environmental signals. Toxicol. Environ. Chem. 2012, 94, 1175–1187. [Google Scholar] [CrossRef]
- Koch, S.; Stroisch, T.; Vorac, J.; Herrmann, N.; Leib, N.; Schnautz, S.; Kirins, H.; Förster, I.; Weighardt, H.; Bieber, T. AhR mediates an anti-inflammatory feedback mechanism in human Langerhans cells involving Fcε RI and IDO. Allergy 2017, 72, 1686–1693. [Google Scholar] [CrossRef]
- Simones, T.; Shepherd, D.M. Consequences of AhR activation in steady-state dendritic cells. Toxicol. Sci. 2011, 119, 293–307. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.; Macchiarulo, A.; Vacca, C. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef]
- Hwang, S.-J.; Hwang, Y.-J.; Yun, M.-O.; Kim, J.-H.; Oh, G.-S.; Park, J.-H. Indoxyl 3-sulfate stimulates Th17 differentiation enhancing phosphorylation of c-Src and STAT3 to worsen experimental autoimmune encephalomyelitis. Toxicol. Lett. 2013, 220, 109–117. [Google Scholar] [CrossRef]
- Ghimire, S.; Matos, C.; Caioni, M.; Weber, D.; Peter, K.; Holler, E.; Kreutz, M.; Renner, K. Indoxyl 3-sulfate inhibits maturation and activation of human monocyte-derived dendritic cells. Immunobiology 2018, 223, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, F.; Coindre, S.; Gardet, M.; Meurisse, F.; Naji, A.; Suganuma, N.; Abi-Rached, L.; Lambotte, O.; Favier, B. Leukocyte immunoglobulin-like receptors in regulating the immune response in infectious diseases: A window of opportunity to pathogen persistence and a sound target in therapeutics. Front. Immunol. 2021, 12, 717998. [Google Scholar] [CrossRef]
- Barroso, A.; Mahler, J.V.; Fonseca-Castro, P.H.; Quintana, F.J. Therapeutic induction of tolerogenic dendritic cells via aryl hydrocarbon receptor signaling. Curr. Opin. Immunol. 2021, 70, 33–39. [Google Scholar] [CrossRef]
- Kado, S.; Chang, W.W.; Chi, A.N.; Wolny, M.; Shepherd, D.M.; Vogel, C.F. Aryl hydrocarbon receptor signaling modifies Toll-like receptor-regulated responses in human dendritic cells. Arch. Toxicol. 2017, 91, 2209–2221. [Google Scholar] [CrossRef]
- Brandstätter, O.; Schanz, O.; Vorac, J.; König, J.; Mori, T.; Maruyama, T.; Korkowski, M.; Haarmann-Stemmann, T.; von Smolinski, D.; Schultze, J.L. Balancing intestinal and systemic inflammation through cell type-specific expression of the aryl hydrocarbon receptor repressor. Sci. Rep. 2016, 6, 26091. [Google Scholar] [CrossRef]
- Jurado-Manzano, B.B.; Zavala-Reyes, D.; Turrubiartes-Martínez, E.A.; Portales-Pérez, D.P.; González-Amaro, R.; Layseca-Espinosa, E. FICZ generates human tDCs that induce CD4+ CD25high Foxp3+ Treg-like cell differentiation. Immunol. Lett. 2017, 190, 84–92. [Google Scholar] [CrossRef]
- Wang, C.; Ye, Z.; Kijlstra, A.; Zhou, Y.; Yang, P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2014, 177, 521–530. [Google Scholar] [CrossRef]
- von Bergwelt-Baildon, M.S.; Popov, A.; Saric, T.; Chemnitz, J.; Classen, S.; Stoffel, M.S.; Fiore, F.; Roth, U.; Beyer, M.; Debey, S. CD25 and indoleamine 2, 3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: Additional mechanisms of T-cell inhibition. Blood 2006, 108, 228–237. [Google Scholar] [CrossRef]
- Driesen, J.; Popov, A.; Schultze, J.L. CD25 as an immune regulatory molecule expressed on myeloid dendritic cells. Immunobiology 2008, 213, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Ciubotariu, R.; Manavalan, J.; Yuan, J.; Colovai, A.I.; Piazza, F.; Lederman, S.; Colonna, M.; Cortesini, R.; Dalla-Favera, R. Tolerization of dendritic cells by TS cells: The crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 2002, 3, 237–243. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Robson, S.; Quintana, F.J. Regulation of the T cell response by CD39. Trends Immunol. 2016, 37, 427–439. [Google Scholar] [CrossRef]
- Silva-Vilches, C.; Ring, S.; Mahnke, K. ATP and its metabolite adenosine as regulators of dendritic cell activity. Front. Immunol. 2018, 9, 2581. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.; Vollmann, E.H.; von Andrian, U.H. Mechanisms and consequences of dendritic cell migration. Immunity 2008, 29, 325–342. [Google Scholar] [CrossRef]
- Casado, F.L.; Singh, K.P.; Gasiewicz, T.A. Aryl hydrocarbon receptor activation in hematopoietic stem/progenitor cells alters cell function and pathway-specific gene modulation reflecting changes in cellular trafficking and migration. Mol. Pharmacol. 2011, 80, 673–682. [Google Scholar] [CrossRef]
- Jin, G.-B.; Moore, A.J.; Head, J.L.; Neumiller, J.J.; Lawrence, B.P. Aryl hydrocarbon receptor activation reduces dendritic cell function during influenza virus infection. Toxicol. Sci. 2010, 116, 514–522. [Google Scholar] [CrossRef]
- Meyers, J.L.; Winans, B.; Kelsaw, E.; Murthy, A.; Gerber, S.; Lawrence, B.P. Environmental cues received during development shape dendritic cell responses later in life. PLoS ONE 2018, 13, e0207007. [Google Scholar] [CrossRef]
- Safe, S.; Jin, U.-h.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl hydrocarbon receptor (AHR) ligands as selective AHR modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Han, H.; Goldsby, J.; Mohankumar, K.; Chapkin, R.S. Aryl hydrocarbon receptor (AhR) ligands as selective AhR modulators: Genomic studies. Curr. Opin. Toxicol. 2018, 11, 10–20. [Google Scholar] [CrossRef]
- Pfeiffer, I.A.; Zinser, E.; Strasser, E.; Stein, M.F.; Dörrie, J.; Schaft, N.; Steinkasserer, A.; Knippertz, I. Leukoreduction system chambers are an efficient, valid, and economic source of functional monocyte-derived dendritic cells and lymphocytes. Immunobiology 2013, 218, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Knippertz, I.; Hesse, A.; Schunder, T.; Kämpgen, E.; Brenner, M.K.; Schuler, G.; Steinkasserer, A.; Nettelbeck, D.M. Generation of human dendritic cells that simultaneously secrete IL-12 and have migratory capacity by adenoviral gene transfer of hCD40L in combination with IFN-γ. J. Immunother. 2009, 32, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Stoner, R.; Maures, T.; Conant, D. Methods and Systems for Guide RNA Design and Use. U.S. Patent 11,345,932, 31 May 2022. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE Guidelines: M inimum I nformation for Publication of Q uantitative Real-Time PCR E xperiments; Oxford University Press: Oxford, UK, 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | Orientation | Sequence |
|---|---|---|
| AHR | Forward | 5′-ACATCACCTACGCCAGTCG-3′ |
| Reverse | 5′-CGCTTGGAAGGATTTGACTTGA-3′ | |
| AHRR | Forward | 5′-GCGCCTCAGTGTCAGTTACC-3′ |
| Reverse | 5′-GAAGCCCAGATAGTCCACGAT-3′ | |
| CYP1A1 | Forward | 5′-TCATCCCCTATTCTTCGCTACC-3′ |
| Reverse | 5′-TCTCCTGACAGTGCTCAATC-3′ | |
| CYP1B1 | Forward | 5′-AACGTACCGGCCACTATCAC-3′ |
| Reverse | 5′-TCACCCATACAAGGCAGACG-3′ | |
| CD83 | Forward | 5′-TGCTGCTGGCTCTGGTTATT-3′ |
| Reverse | 5′-TGTGAGGAGTCACTAGCCCT-3′ | |
| CD80 | Forward | 5′-CCATCCAAGTGTCCATACCTC-3′ |
| Reverse | 5′-GCCAGCTCTTCAACAGAAAC-3′ | |
| CD25 (IL2RA) | Forward | 5′-ACTTCCTGCCTCGTCAC-3′ |
| Reverse | 5′-TCTACTCTTCCTCTGTCTCCG-3′ | |
| CCR7 | Forward | 5′-GCTCTCCTTGTCATTTTCCAG-3′ |
| Reverse | 5′-GCTTTAAAGTTCCGCACGTC-3′ | |
| CD39 | Forward | 5′-AGAGGAAGGTGCCTATGGCT-3′ |
| Reverse | 5′-TGGGGACTCGATAGTCTGGTT-3′ | |
| CD73 | Forward | 5′-AGTACCAGGGCACTATCTGGT-3′ |
| Reverse | 5′-TGAGGAGTGGCTCGATCAGT-3′ | |
| ILT3 | Forward | 5′-CTTCAGCTCACACGGCTTCT-3′ |
| Reverse | 5′-ACTGACCCTGTAGGCATGAG-3′ | |
| ILT4 | Forward | 5′-CTCAACTCCGACCCCTACCT-3′ |
| Reverse | 5′-AAGATGCCGATCACAACCCC-3′ | |
| ILT5 | Forward | 5′-GACAGAGCCCAAGGACAG-3′ |
| Reverse | 5′-GGGTCTTCATCGTGTGGG-3′ | |
| CHCHD2 | Forward | 5′-CACATTGGGTCACGCCATTA-3′ |
| Reverse | 5′-GCTTGATGTCACCCTGGTTCT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeghi Shermeh, A.; Royzman, D.; Kuhnt, C.; Draßner, C.; Stich, L.; Steinkasserer, A.; Knippertz, I.; Wild, A.B. Differential Modulation of Dendritic Cell Biology by Endogenous and Exogenous Aryl Hydrocarbon Receptor Ligands. Int. J. Mol. Sci. 2023, 24, 7801. https://doi.org/10.3390/ijms24097801
Sadeghi Shermeh A, Royzman D, Kuhnt C, Draßner C, Stich L, Steinkasserer A, Knippertz I, Wild AB. Differential Modulation of Dendritic Cell Biology by Endogenous and Exogenous Aryl Hydrocarbon Receptor Ligands. International Journal of Molecular Sciences. 2023; 24(9):7801. https://doi.org/10.3390/ijms24097801
Chicago/Turabian StyleSadeghi Shermeh, Atefeh, Dmytro Royzman, Christine Kuhnt, Christina Draßner, Lena Stich, Alexander Steinkasserer, Ilka Knippertz, and Andreas B. Wild. 2023. "Differential Modulation of Dendritic Cell Biology by Endogenous and Exogenous Aryl Hydrocarbon Receptor Ligands" International Journal of Molecular Sciences 24, no. 9: 7801. https://doi.org/10.3390/ijms24097801