
Molecular and Physiological Determinants of Amyotrophic Lateral Sclerosis: What the DJ-1 Protein Teaches Us

{kind=link}
{kind=link}
Abstract
:1. Introduction
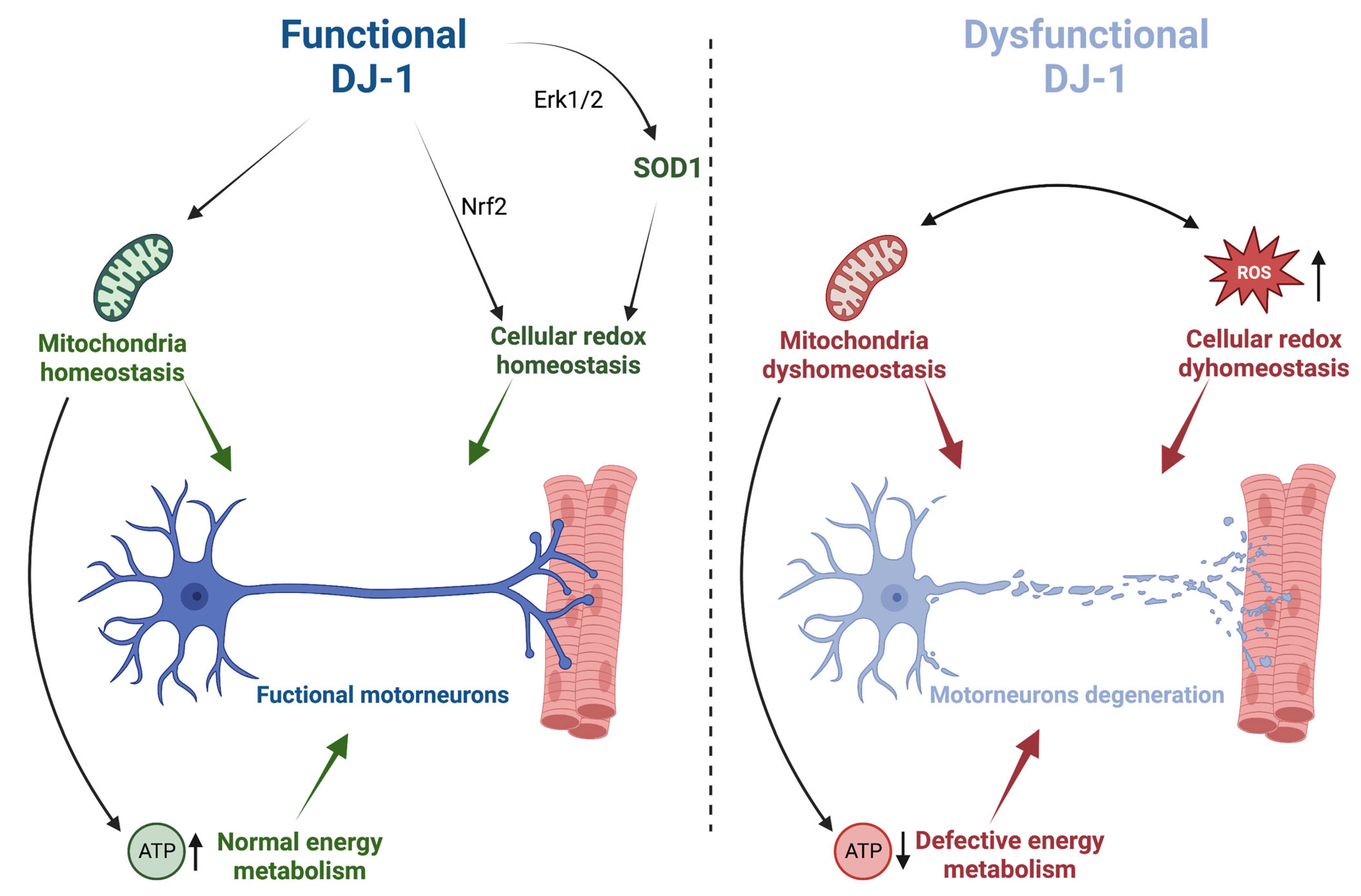
2. DJ-1 Protection against Oxidative Stress and Mitochondrial Dysfunction: Implications for ALS
3. DJ-1 Involvement in Energy Metabolism: Implications for ALS
4. DJ-1 Involvement in Hypoxia Response: Implications for ALS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.S.A.; Antonyuk, S.V.; Hasnain, S.S. The Biophysics of Superoxide Dismutase-1 and Amyotrophic Lateral Sclerosis. Q. Rev. Biophys. 2019, 52, e12. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. Engl. 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.H.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. CuII(Atsm) Improves the Neurological Phenotype and Survival of SOD1G93A Mice and Selectively Increases Enzymatically Active SOD1 in the Spinal Cord. Sci. Rep. 2017, 7, 42292. [Google Scholar] [CrossRef]
- Williams, J.R.; Trias, E.; Beilby, P.R.; Lopez, N.I.; Labut, E.M.; Bradford, C.S.; Roberts, B.R.; McAllum, E.J.; Crouch, P.J.; Rhoads, T.W.; et al. Copper Delivery to the CNS by CuATSM Effectively Treats Motor Neuron Disease in SOD(G93A) Mice Co-Expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 2016, 89, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Lim, N.K.H.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral Treatment with Cu(II)(Atsm) Increases Mutant SOD1 in Vivo but Protects Motor Neurons and Improves the Phenotype of a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef]
- Trist, B.G.; Genoud, S.; Roudeau, S.; Rookyard, A.; Abdeen, A.; Cottam, V.; Hare, D.J.; White, M.; Altvater, J.; Fifita, J.A.; et al. Altered SOD1 Maturation and Post-Translational Modification in Amyotrophic Lateral Sclerosis Spinal Cord. Brain 2022, 145, 3108–3130. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP Mutations in Individuals with Sporadic and Familial Amyotrophic Lateral Sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.C.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP Mutations in Amyotrophic Lateral Sclerosis with TDP-43 Neuropathology: A Genetic and Histopathological Analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Salado, I.G.; De La Encarnación, A.; Pérez, D.I.; Moreno, F.; Gil, C.; De Munain, A.L.; Martínez, A.; Martín-Requero, Á. Targeting TDP-43 Phosphorylation by Casein Kinase-1δ Inhibitors: A Novel Strategy for the Treatment of Frontotemporal Dementia. Mol. Neurodegener. 2016, 11, 36. [Google Scholar] [CrossRef]
- Annesi, G.; Savettieri, G.; Pugliese, P.; D’Amelio, M.; Tarantino, P.; Ragonese, P.; La Bella, V.; Piccoli, T.; Civitelli, D.; Annesi, F.; et al. DJ-1 Mutations and Parkinsonism-Dementia-Amyotrophic Lateral Sclerosis Complex. Ann. Neurol. 2005, 58, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent Findings on the Physiological Function of DJ-1: Beyond Parkinson’s Disease. Neurobiol. Dis. 2017, 108, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hanagasi, H.A.; Giri, A.; Kartal, E.; Guven, G.; Bilgiç, B.; Hauser, A.K.; Emre, M.; Heutink, P.; Basak, N.; Gasser, T.; et al. A Novel Homozygous DJ1 Mutation Causes Parkinsonism and ALS in a Turkish Family. Park. Relat. Disord. 2016, 29, 117–120. [Google Scholar] [CrossRef]
- Yamashita, S.; Mori, A.; Kimura, E.; Mita, S.; Maeda, Y.; Hirano, T.; Uchino, M. DJ-1 Forms Complexes with Mutant SOD1 and Ameliorates Its Toxicity. J. Neurochem. 2010, 113, 860–870. [Google Scholar] [CrossRef]
- Knippenberg, S.; Sipos, J.; Thau-Habermann, N.; Körner, S.; Rath, K.J.; Dengler, R.; Petri, S. Altered Expression of DJ-1 and PINK1 in Sporadic ALS and in the SOD1(G93A) ALS Mouse Model. J. Neuropathol. Exp. Neurol. 2013, 72, 1052–1061. [Google Scholar] [CrossRef]
- Lev, N.; Barhum, Y.; Lotan, I.; Steiner, I.; Offen, D. DJ-1 Knockout Augments Disease Severity and Shortens Survival in a Mouse Model of ALS. PLoS ONE 2015, 10, e0117190. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, Z.F.; Lei, R.X.; Wang, S.; Zhuang, Y.; Liu, A.C.; Wu, Y.; Chen, J.; Tang, J.C.; Pan, M.X.; et al. DJ-1 Suppresses Cytoplasmic TDP-43 Aggregation in Oxidative Stress-Induced Cell Injury. J. Alzheimers Dis. 2018, 66, 1001–1014. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate Up-Regulates the DJ-1 Protein and Protects Neurons in Cell Culture and in Animal Models of Parkinson Disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef]
- Yang, R.X.; Lei, J.; Wang, B.D.; Feng, D.Y.; Huang, L.; Li, Y.Q.; Li, T.; Zhu, G.; Li, C.; Lu, F.F.; et al. Pretreatment with Sodium Phenylbutyrate Alleviates Cerebral Ischemia/Reperfusion Injury by Upregulating DJ-1 Protein. Front. Neurol. 2017, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-Cell-Autonomous Pathogenic Mechanisms in Amyotrophic Lateral Sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Krüger, R. The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- De Lazzari, F.; Bisaglia, M. DJ-1 as a Deglycating Enzyme: A Unique Function to Explain a Multifaceted Protein? Neural Regen. Res. 2017, 12, 1797–1798. [Google Scholar] [CrossRef] [PubMed]
- De Lazzari, F.; Prag, H.A.; Gruszczyk, A.V.; Whitworth, A.J.; Bisaglia, M. DJ-1: A Promising Therapeutic Candidate for Ischemia-Reperfusion Injury. Redox Biol. 2021, 41, 101884. [Google Scholar] [CrossRef]
- De Lazzari, F.; Agostini, F.; Plotegher, N.; Sandre, M.; Greggio, E.; Megighian, A.; Bubacco, L.; Sandrelli, F.; Whitworth, A.J.; Bisaglia, M. DJ-1 Promotes Energy Balance by Regulating Both Mitochondrial and Autophagic Homeostasis. Neurobiol. Dis. 2023, 176, 105941. [Google Scholar] [CrossRef]
- De Lazzari, F.; Agostini, F.; Doni, D.; Malacrida, S.; Zordan, M.A.; Costantini, P.; Bubacco, L.; Sandrelli, F.; Bisaglia, M. DJ-1 and SOD1 Act Independently in the Protection against Anoxia in Drosophila Melanogaster. Antioxidants 2022, 11, 1527. [Google Scholar] [CrossRef]
- Muthukumaran, K.; Smith, J.; Jasra, H.; Sikorska, M.; Sandhu, J.K.; Cohen, J.; Lopatin, D.; Pandey, S. Genetic Susceptibility Model of Parkinson’s Disease Resulting from Exposure of DJ-1 Deficient Mice to MPTP: Evaluation of Neuroprotection by Ubisol-Q10. J. Parkinsons Dis. 2014, 4, 523–530. [Google Scholar] [CrossRef]
- Lavara-Culebras, E.; Paricio, N. Drosophila DJ-1 Mutants Are Sensitive to Oxidative Stress and Show Reduced Lifespan and Motor Deficits. Gene 2007, 400, 158–165. [Google Scholar] [CrossRef]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 Mutants Are Selectively Sensitive to Environmental Toxins Associated with Parkinson’s Disease. Curr. Biol. 2005, 15, 1572–1577. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-Deficient Mice to 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyrindine (MPTP) and Oxidative Stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [PubMed]
- Batelli, S.; Invernizzi, R.W.; Negro, A.; Calcagno, E.; Rodilossi, S.; Forloni, G.; Albani, D. The Parkinson’s Disease-Related Protein DJ-1 Protects Dopaminergic Neurons in Vivo and Cultured Cells from Alpha-Synuclein and 6-Hydroxydopamine Toxicity. Neurodegener. Dis. 2015, 15, 13–23. [Google Scholar] [CrossRef]
- Martinat, C.; Shendelman, S.; Jonason, A.; Leete, T.; Beal, M.F.; Yang, L.; Floss, T.; Abeliovich, A. Sensitivity to Oxidative Stress in DJ-1-Deficient Dopamine Neurons: An ES- Derived Cell Model of Primary Parkinsonism. PLoS Biol. 2004, 2, e327. [Google Scholar] [CrossRef] [PubMed]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Takahashi, K.; Ariga, H. DJ-1 Has a Role in Antioxidative Stress to Prevent Cell Death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A. The Role of Cysteine Oxidation in DJ-1 Function and Dysfunction. Antioxid. Redox Signal. 2011, 15, 111–122. [Google Scholar] [CrossRef]
- Huang, M.; Chen, S. DJ-1 in Neurodegenerative Diseases: Pathogenesis and Clinical Application. Prog. Neurobiol. 2021, 204, 102114. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J.; Chen, S.; Wang, Y.; Cao, L.; Zhang, Y.; Kang, W.; Li, H.; Gui, Y.; Chen, S.; et al. DJ-1 Modulates the Expression of Cu/Zn-Superoxide Dismutase-1 through the Erk1/2-Elk1 Pathway in Neuroprotection. Ann. Neurol. 2011, 70, 591–599. [Google Scholar] [CrossRef]
- Yan, Y.F.; Yang, W.J.; Xu, Q.; Chen, H.P.; Huang, X.S.; Qiu, L.Y.; Liao, Z.P.; Huang, Q.R. DJ-1 Upregulates Anti-Oxidant Enzymes and Attenuates Hypoxia/Re-Oxygenation-Induced Oxidative Stress by Activation of the Nuclear Factor Erythroid 2-like 2 Signaling Pathway. Mol. Med. Rep. 2015, 12, 4734–4742. [Google Scholar] [CrossRef]
- Yan, Y.F.; Chen, H.P.; Huang, X.S.; Qiu, L.Y.; Liao, Z.P.; Huang, Q.R. DJ-1 Mediates the Delayed Cardioprotection of Hypoxic Preconditioning Through Activation of Nrf2 and Subsequent Upregulation of Antioxidative Enzymes. J. Cardiovasc. Pharmacol. 2015, 66, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.Y. DJ-1, a Cancer- and Parkinson’s Disease-Associated Protein, Stabilizes the Antioxidant Transcriptional Master Regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Arslanbaeva, L.; Bisaglia, M. Activation of the Nrf2 Pathway as a Therapeutic Strategy for ALS Treatment. Molecules 2022, 27, 1471. [Google Scholar] [CrossRef]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The NRF2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Bono, S.; Feligioni, M.; Corbo, M. Impaired Antioxidant KEAP1-NRF2 System in Amyotrophic Lateral Sclerosis: NRF2 Activation as a Potential Therapeutic Strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Villegas, J.; Ferraiuolo, L.; Mead, R.J.; Shaw, P.J.; Cuadrado, A.; Rojo, A.I. NRF2 as a Therapeutic Opportunity to Impact in the Molecular Roadmap of ALS. Free Radic. Biol. Med. 2021, 173, 125–141. [Google Scholar] [CrossRef]
- Björkblom, B.; Adilbayeva, A.; Maple-Grødem, J.; Piston, D.; Ökvist, M.; Xu, X.M.; Brede, C.; Larsen, J.P.; Møller, S.G. Parkinson Disease Protein DJ-1 Binds Metals and Protects against Metal-Induced Cytotoxicity. J. Biol. Chem. 2013, 288, 22809–22820. [Google Scholar] [CrossRef]
- Puno, M.R.; Patel, N.A.; Møller, S.G.; Robinson, C.V.; Moody, P.C.E.; Odell, M. Structure of Cu(I)-Bound DJ-1 Reveals a Biscysteinate Metal Binding Site at the Homodimer Interface: Insights into Mutational Inactivation of DJ-1 in Parkinsonism. J. Am. Chem. Soc. 2013, 135, 15974–15977. [Google Scholar] [CrossRef]
- Girotto, S.; Cendron, L.; Bisaglia, M.; Tessari, I.; Mammi, S.; Zanotti, G.; Bubacco, L. DJ-1 Is a Copper Chaperone Acting on SOD1 Activation. J. Biol. Chem. 2014, 289, 10887–10899. [Google Scholar] [CrossRef]
- Xu, X.M.; Lin, H.; Maple, J.; Björkblom, B.; Alves, G.; Larsen, J.P.; Møller, S.G. The Arabidopsis DJ-1a Protein Confers Stress Protection through Cytosolic SOD Activation. J. Cell Sci. 2010, 123, 1644–1651. [Google Scholar] [CrossRef]
- Barbieri, L.; Luchinat, E.; Banci, L. Intracellular Metal Binding and Redox Behavior of Human DJ-1. J. Biol. Inorg. Chem. 2018, 23, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, D.; Calì, T.; Negro, A.; Brini, M. The Parkinson Disease-Related Protein DJ-1 Counteracts Mitochondrial Impairment Induced by the Tumour Suppressor Protein P53 by Enhancing Endoplasmic Reticulum-Mitochondria Tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Park, J.H.; Kim, S.J.; Seo, K.S.; Han, J.S.; Lee, S.H.; Kim, J.M.; Park, J.I.; Park, S.K.; Lim, K.; et al. DJ-1 Null Dopaminergic Neuronal Cells Exhibit Defects in Mitochondrial Function and Structure: Involvement of Mitochondrial Complex I Assembly. PLoS ONE 2012, 7, e32629. [Google Scholar] [CrossRef]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 Acts in Parallel to the PINK1/Parkin Pathway to Control Mitochondrial Function and Autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.C.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s Disease-Linked Gene DJ-1 Perturbs Mitochondrial Dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced Basal Autophagy and Impaired Mitochondrial Dynamics Due to Loss of Parkinson’s Disease-Associated Protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Primiani, C.T.; Cookson, M.R. The Effects of Variants in the Parkin, PINK1, and DJ-1 Genes along with Evidence for Their Pathogenicity. Curr. Protein Pept. Sci. 2017, 18, 702–714. [Google Scholar] [CrossRef]
- Imberechts, D.; Kinnart, I.; Wauters, F.; Terbeek, J.; Manders, L.; Wierda, K.; Eggermont, K.; Madeiro, R.F.; Sue, C.; Verfaillie, C.; et al. DJ-1 Is an Essential Downstream Mediator in PINK1/Parkin-Dependent Mitophagy. Brain 2022, 145, 4368–4384. [Google Scholar] [CrossRef]
- Ozawa, K.; Tsumoto, H.; Miura, Y.; Yamaguchi, J.; Iguchi-Ariga, S.M.M.; Sakuma, T.; Yamamoto, T.; Uchiyama, Y. DJ-1 Is Indispensable for the S-Nitrosylation of Parkin, Which Maintains Function of Mitochondria. Sci. Rep. 2020, 10, 4377. [Google Scholar] [CrossRef]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 Is Critical for Mitochondrial Function and Rescues PINK1 Loss of Function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Soriano, M.E.; Brini, M. A New Split-GFP-Based Probe Reveals DJ-1 Translocation into the Mitochondrial Matrix to Sustain ATP Synthesis upon Nutrient Deprivation. Hum. Mol. Genet. 2015, 24, 1045–1060. [Google Scholar] [CrossRef]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial Localization of DJ-1 Leads to Enhanced Neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s Disease Protein DJ-1 Is Neuroprotective Due to Cysteine-Sulfinic Acid-Driven Mitochondrial Localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Soares, P.; Silva, C.; Chavarria, D.; Silva, F.S.G.; Oliveira, P.J.; Borges, F. Drug Discovery and Amyotrophic Lateral Sclerosis: Emerging Challenges and Therapeutic Opportunities. Ageing Res. Rev. 2023, 83, 101790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
- Solana-Manrique, C.; Sanz, F.J.; Ripollés, E.; Bañó, M.C.; Torres, J.; Muñoz-Soriano, V.; Paricio, N. Enhanced Activity of Glycolytic Enzymes in Drosophila and Human Cell Models of Parkinson’s Disease Based on DJ-1 Deficiency. Free Radic. Biol. Med. 2020, 158, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Mamais, A.; Conti, M.M.; Primiani, C.T.; Kumaran, R.; Dillman, A.A.; Langston, R.G.; Beilina, A.; Garcia, J.H.; Diaz-Ruiz, A.; et al. Hexokinases Link DJ-1 to the PINK1/Parkin Pathway. Mol. Neurodegener. 2017, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Jimenez-Blasco, D.; Fernandez, E.; Almeida, A.; Bolaños, J.P. DJ1 Represses Glycolysis and Cell Proliferation by Transcriptionally Up-Regulating Pink1. Biochem. J. 2015, 467, 303–310. [Google Scholar] [CrossRef]
- Xu, M.; Wu, H.; Li, M.; Wen, Y.; Yu, C.; Xia, L.; Xia, Q.; Kong, X. DJ-1 Deficiency Protects Hepatic Steatosis by Enhancing Fatty Acid Oxidation in Mice. Int. J. Biol. Sci. 2018, 14, 1892–1900. [Google Scholar] [CrossRef]
- Wu, R.; Liu, X.M.; Sun, J.G.; Chen, H.; Ma, J.; Dong, M.; Peng, S.; Wang, J.Q.; Ding, J.Q.; Li, D.H.; et al. DJ-1 Maintains Energy and Glucose Homeostasis by Regulating the Function of Brown Adipose Tissue. Cell Discov. 2017, 3, 16054. [Google Scholar] [CrossRef]
- Kim, J.M.; Jang, H.J.; Choi, S.Y.; Park, S.A.; Kim, I.S.; Yang, Y.R.; Lee, Y.H.; Ryu, S.H.; Suh, P.G. DJ-1 Contributes to Adipogenesis and Obesity-Induced Inflammation. Sci. Rep. 2014, 4, 4805. [Google Scholar] [CrossRef] [PubMed]
- Solana-Manrique, C.; Sanz, F.J.; Torregrosa, I.; Palomino-Schätzlein, M.; Hernández-Oliver, C.; Pineda-Lucena, A.; Paricio, N. Metabolic Alterations in a Drosophila Model of Parkinson’s Disease Based on DJ-1 Deficiency. Cells 2022, 11, 331. [Google Scholar] [CrossRef]
- Vanweert, F.; Schrauwen, P.; Phielix, E. Role of Branched-Chain Amino Acid Metabolism in the Pathogenesis of Obesity and Type 2 Diabetes-Related Metabolic Disturbances BCAA Metabolism in Type 2 Diabetes. Nutr. Diabetes 2022, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Guillot, S.J.; Bolborea, M.; Dupuis, L. Dysregulation of Energy Homeostasis in Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 2021, 34, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Li, R.; Kirk, S.E.; Tefera, T.W.; Xie, T.Y.; Tracey, T.J.; Kelk, D.; Wimberger, E.; Garton, F.C.; Roberts, L.; et al. Altered Skeletal Muscle Glucose-Fatty Acid Flux in Amyotrophic Lateral Sclerosis. Brain Commun. 2020, 2, fcaa154. [Google Scholar] [CrossRef]
- Szelechowski, M.; Amoedo, N.; Obre, E.; Léger, C.; Allard, L.; Bonneu, M.; Claverol, S.; Lacombe, D.; Oliet, S.; Chevallier, S.; et al. Metabolic Reprogramming in Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 3953. [Google Scholar] [CrossRef]
- Raman, R.; Allen, S.P.; Goodall, E.F.; Kramer, S.; Ponger, L.L.; Heath, P.R.; Milo, M.; Hollinger, H.C.; Walsh, T.; Highley, J.R.; et al. Gene Expression Signatures in Motor Neurone Disease Fibroblasts Reveal Dysregulation of Metabolism, Hypoxia-Response and RNA Processing Functions. Neuropathol. Appl. Neurobiol. 2015, 41, 201–226. [Google Scholar] [CrossRef]
- Lederer, C.W.; Torrisi, A.; Pantelidou, M.; Santama, N.; Cavallaro, S. Pathways and Genes Differentially Expressed in the Motor Cortex of Patients with Sporadic Amyotrophic Lateral Sclerosis. BMC Genom. 2007, 8, 26. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; René, F.; Gonzalez De Aguilar, J.L.; Loeffler, J.P. Evidence for Defective Energy Homeostasis in Amyotrophic Lateral Sclerosis: Benefit of a High-Energy Diet in a Transgenic Mouse Model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H.M. A High-Fat Jelly Diet Restores Bioenergetic Balance and Extends Lifespan in the Presence of Motor Dysfunction and Lumbar Spinal Cord Motor Neuron Loss in TDP-43A315T Mutant C57BL6/J Mice. Dis. Model. Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef]
- Wills, A.M.; Hubbard, J.; Macklin, E.A.; Glass, J.; Tandan, R.; Simpson, E.P.; Brooks, B.; Gelinas, D.; Mitsumoto, H.; Mozaffar, T.; et al. Hypercaloric Enteral Nutrition in Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Phase 2 Trial. Lancet 2014, 383, 2065–2072. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A Ketogenic Diet as a Potential Novel Therapeutic Intervention in Amyotrophic Lateral Sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-Chain Fatty Acids, Beta-Hydroxybutyric Acid and Genetic Modulation of the Carnitine Shuttle Are Protective in a Drosophila Model of ALS Based on TDP-43. Front. Mol. Neurosci. 2018, 11, 182. [Google Scholar] [CrossRef]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases-New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef] [PubMed]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xue, Y.; Li, Y.; Gao, S.; Peng, L.; Zhao, Y.; Yu, S. DJ-1 Protein Inhibits Apoptosis in Cerebral Ischemia by Regulating the Notch1 and Nuclear Factor Erythroid2-Related Factor 2 Signaling Pathways. Neuroscience 2022, 504, 33–46. [Google Scholar] [CrossRef]
- Peng, L.; Zhao, Y.; Li, Y.; Zhou, Y.; Li, L.; Lei, S.; Yu, S.; Zhao, Y. Effect of DJ-1 on the Neuroprotection of Astrocytes Subjected to Cerebral Ischemia/Reperfusion Injury. J. Mol. Med. 2019, 97, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Molcho, L.; Ben-Zur, T.; Barhum, Y.; Offen, D. DJ-1 Based Peptide, ND-13, Promote Functional Recovery in Mouse Model of Focal Ischemic Injury. PLoS ONE 2018, 13, e0192954. [Google Scholar] [CrossRef]
- Aleyasin, H.; Rousseaux, M.W.C.; Phillips, M.; Kim, R.H.; Bland, R.J.; Callaghan, S.; Slack, R.S.; During, M.J.; Mak, T.W.; Park, D.S. The Parkinson’s Disease Gene DJ-1 Is Also a Key Regulator of Stroke-Induced Damage. Proc. Natl. Acad. Sci. USA 2007, 104, 18748–18753. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Kim, J.J.; Wolfson, D.W.; Cho, H.C.; Husain, A.; Naqvi, N.; Chin, L.S.; Li, L.; et al. DJ-1 Protects the Heart against Ischemia-Reperfusion Injury by Regulating Mitochondrial Fission. J. Mol. Cell. Cardiol. 2016, 97, 56–66. [Google Scholar] [CrossRef]
- Dongworth, R.K.; Mukherjee, U.A.; Hall, A.R.; Astin, R.; Ong, S.B.; Yao, Z.; Dyson, A.; Szabadkai, G.; Davidson, S.M.; Yellon, D.M.; et al. DJ-1 Protects against Cell Death Following Acute Cardiac Ischemia-Reperfusion Injury. Cell Death Dis. 2014, 5, e1082. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Kitamura, Y.; Inden, M.; Takata, K.; Taniguchi, T.; Morikawa, S.; Morita, M.; Inubushi, T.; Tooyama, I.; Taira, T.; et al. DJ-1 Protects against Neurodegeneration Caused by Focal Cerebral Ischemia and Reperfusion in Rats. J. Cereb. Blood Flow. Metab. 2008, 28, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, T.; Peng, L.; Li, Y.; Zhao, Y.; Yu, S. Attenuation of Inflammation by DJ-1 May Be a Drug Target for Cerebral Ischemia-Reperfusion Injury. Neurochem. Res. 2021, 46, 1470–1479. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Ren, J.M.; Liu, H.R.; Zhou, T.T.; Wang, X.Y.; Liu, S.; Chen, H.P. DJ-1 Activates the AMPK/MTOR Pathway by Binding RACK1 to Induce Autophagy and Protect the Myocardium from Ischemia/Hypoxia Injury. Biochem. Biophys. Res. Commun. 2022, 637, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Gallinat, A.; Mendieta, G.; Vilahur, G.; Padró, T.; Badimon, L. DJ-1 Administration Exerts Cardioprotection in a Mouse Model of Acute Myocardial Infarction. Front. Pharmacol. 2022, 13, 1002755. [Google Scholar] [CrossRef]
- Yu, H.H.; Xu, Q.; Chen, H.P.; Wang, S.; Huang, X.S.; Huang, Q.R.; He, M. Stable Overexpression of DJ-1 Protects H9c2 Cells against Oxidative Stress under a Hypoxia Condition. Cell Biochem. Funct. 2013, 31, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 Is an Important Mediator of Hypoxia-Induced Cellular Responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef]
- Parsanejad, M.; Zhang, Y.; Qu, D.; Irrcher, I.; Rousseaux, M.W.C.; Aleyasin, H.; Kamkar, F.; Callaghan, S.; Slack, R.S.; Mak, T.W.; et al. Regulation of the VHL/HIF-1 Pathway by DJ-1. J. Neurosci. 2014, 34, 8043–8050. [Google Scholar] [CrossRef]
- Zheng, H.; Zhou, C.; Lu, X.; Liu, Q.; Liu, M.; Chen, G.; Chen, W.; Wang, S.; Qiu, Y. DJ-1 Promotes Survival of Human Colon Cancer Cells under Hypoxia by Modulating HIF-1α Expression through the PI3K-AKT Pathway. Cancer Manag. Res. 2018, 10, 4615–4629. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Lassalle, P.; Perez, T.; De Seze, J.; Brunaud-Danel, V.; Destée, A.; Tonnel, A.B.; Just, N. Low Levels of the Vascular Endothelial Growth Factor in CSF from Early ALS Patients. Neurology 2004, 62, 2127–2129. [Google Scholar] [CrossRef]
- Just, N.; Moreau, C.; Lassalle, P.; Gosset, P.; Perez, T.; Brunaud-Danel, V.; Wallaert, B.; Destée, A.; Defebvre, L.; Tonnel, A.B.; et al. High Erythropoietin and Low Vascular Endothelial Growth Factor Levels in Cerebrospinal Fluid from Hypoxemic ALS Patients Suggest an Abnormal Response to Hypoxia. Neuromuscul. Disord. 2007, 17, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Devos, D.; Brunaud-Danel, V.; Defebvre, L.; Perez, T.; Destée, A.; Tonnel, A.B.; Lassalle, P.; Just, N. Paradoxical Response of VEGF Expression to Hypoxia in CSF of Patients with ALS. J. Neurol. Neurosurg. Psychiatry 2006, 77, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Gosset, P.; Kluza, J.; Brunaud-Danel, V.; Lassalle, P.; Marchetti, P.; Defebvre, L.; Destée, A.; Devos, D. Deregulation of the Hypoxia Inducible Factor-1α Pathway in Monocytes from Sporadic Amyotrophic Lateral Sclerosis Patients. Neuroscience 2011, 172, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a Therapy for Mitochondrial Disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective Targeting of a Redox-Active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.J.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective Effects of the Mitochondria-Targeted Antioxidant MitoQ in a Model of Inherited Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef]
- Agostini, F.; Masato, A.; Bubacco, L.; Bisaglia, M. Metformin Repurposing for Parkinson Disease Therapy: Opportunities and Challenges. Int. J. Mol. Sci. 2021, 23, 398. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandrelli, F.; Bisaglia, M. Molecular and Physiological Determinants of Amyotrophic Lateral Sclerosis: What the DJ-1 Protein Teaches Us. Int. J. Mol. Sci. 2023, 24, 7674. https://doi.org/10.3390/ijms24087674
Sandrelli F, Bisaglia M. Molecular and Physiological Determinants of Amyotrophic Lateral Sclerosis: What the DJ-1 Protein Teaches Us. International Journal of Molecular Sciences. 2023; 24(8):7674. https://doi.org/10.3390/ijms24087674
Chicago/Turabian StyleSandrelli, Federica, and Marco Bisaglia. 2023. "Molecular and Physiological Determinants of Amyotrophic Lateral Sclerosis: What the DJ-1 Protein Teaches Us" International Journal of Molecular Sciences 24, no. 8: 7674. https://doi.org/10.3390/ijms24087674