
Biochemical Screening for Fetal Trisomy 21: Pathophysiology of Maternal Serum Markers and Involvement of the Placenta
 , , , ,
, , , ,
Abstract
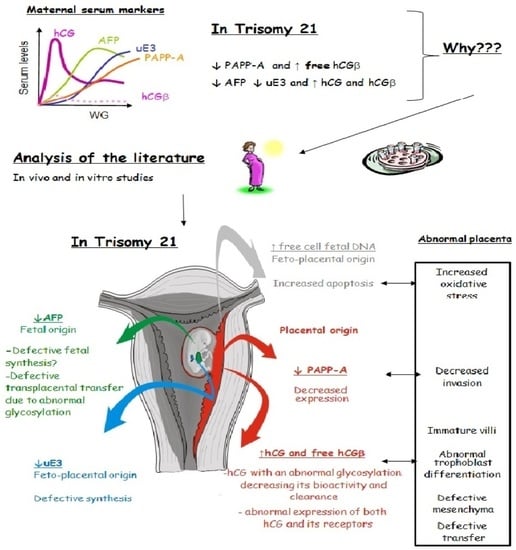
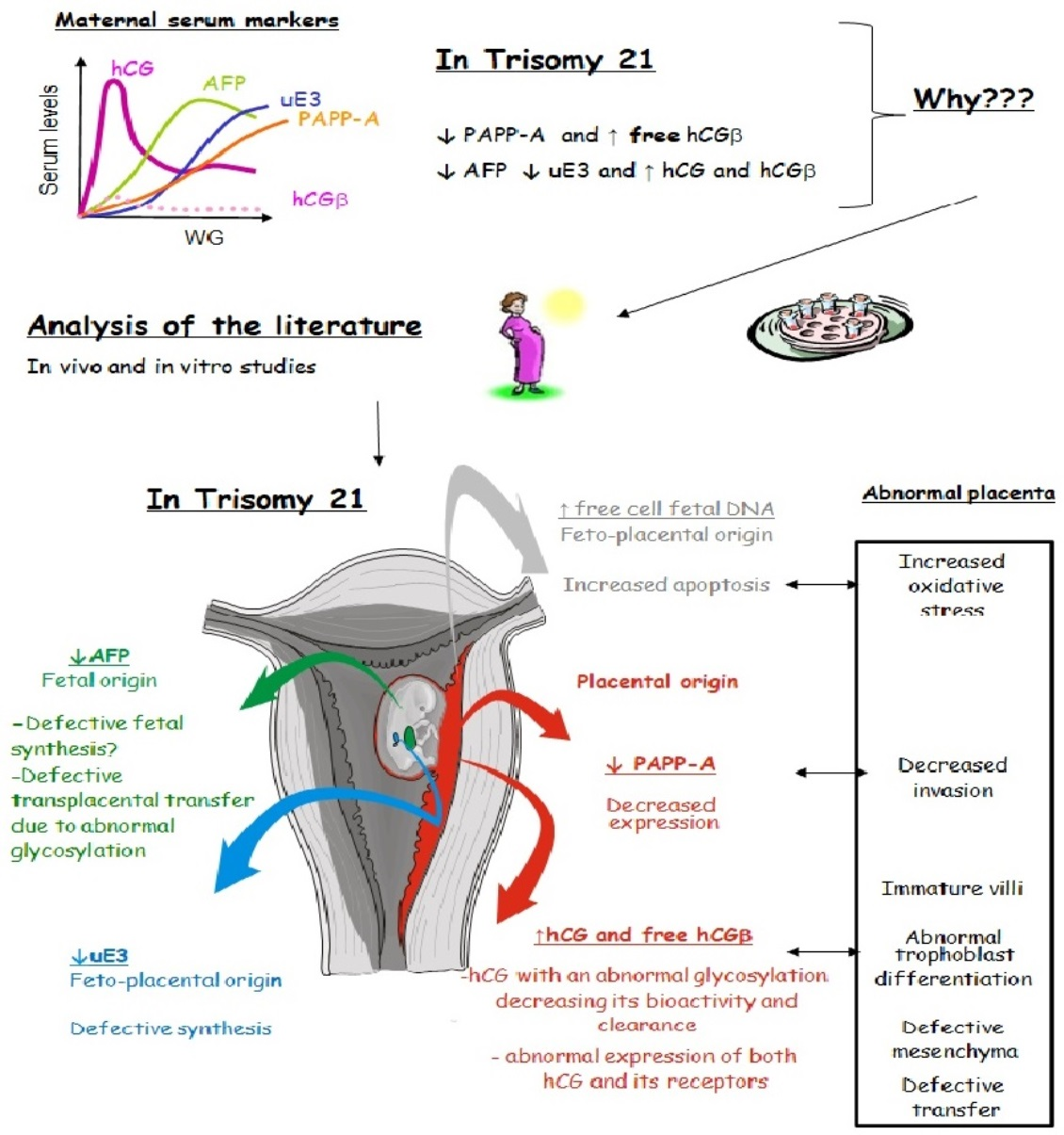
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Soluble Markers
2.1. Markers of Placental Origin
2.1.1. Human Chorionic Gonadotropin
2.1.2. Pregnancy-Associated Plasmatic Protein A
2.1.3. Inhibin A
2.2. Markers of Fetal Origin: The Alpha-Fetoprotein
2.3. Markers of Feto–Placental Origin: The Unconjugated Estriol
3. Cellular Markers and Placenta
3.1. Cellular Markers and Cell-Free Fetal DNA
3.2. Placenta and Trophoblastic Villi in Trisomy 21
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Roizen, N.; Patterson, D. Down’s syndrome. Lancet 2003, 361, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, J.; Turpin, R.; Gautier, M. Chromosomic diagnosis of mongolism. Arch. Fr. Pediatr. 1959, 16, 962–963. [Google Scholar] [PubMed]
- Antonarakis, S.; Lyle, R.; Dermitzakis, E.; Reymond, A.; Deutsch, S. Chromosome 21 and Down syndrome: From genomics to pathophysiology. Nat. Rev. Genet. 2004, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Penrose, L. The relative effects of paternal and maternal age in mongolism. J. Genet. 1933, 27, 219–224. [Google Scholar] [CrossRef]
- Benn, P. Advances in prenatal screening for Down syndrome: I. General principles and second trimester testing. Clin. Chim. Acta 2002, 323, 1–16. [Google Scholar] [CrossRef]
- Snijders, R.; Noble, P.; Sebire, N.; Souka, A.; Nicolaides, K. UK multicentre project on assessment of risk of trisomy 21 by maternal age and fetal nuchal-translucency thickness at 10–14 weeks of gestation. Fetal Medicine Foundation First Trimester Screening Group. Lancet 1998, 352, 343–346. [Google Scholar] [CrossRef]
- Bogart, M.; Pandian, M.; Jones, O. Abnormal maternal serum chorionic gonadotropin levels in pregnancies with fetal chromosome abnormalities. Prenat. Diagn. 1987, 7, 623–630. [Google Scholar] [CrossRef]
- Heyl, P.; Miller, W.; Canick, J. Maternal serum screening for aneuploid pregnancy by alpha-fetoprotein, hCG, and unconjugated estriol. Obs. Gynecol. 1990, 76, 1025–1031. [Google Scholar]
- Merkatz, I.; Nitowsky, H.; Macri, J.; Johnson, W. An association between low maternal serum alpha-fetoprotein and fetal chromosomal abnormalities. Am. J. Obs. Gynecol. 1984, 148, 886–894. [Google Scholar] [CrossRef]
- Canick, J.; Knight, G.; Palomaki, G.; Haddow, J.; Cuckle, H.; Wald, N. Low second trimester maternal serum unconjugated oestriol in pregnancies with Down’s syndrome. Br. J. Obstet. Gynaecol. 1988, 95, 330–333. [Google Scholar] [CrossRef]
- Aitken, D.; Wallace, E.; Crossley, J.; Swanston, I.; van Pareren, Y.; van Maarle, M.; Groome, N.; Macri, J.; Connor, J. Dimeric inhibin A as a marker for Down’s syndrome in early pregnancy. N. Engl. J. Med. 1996, 334, 1231–1236. [Google Scholar] [CrossRef]
- Muller, F.; Cuckle, H.; Teisner, B.; Grudzinskas, J. Serum PAPP–A levels are depressed in women with fetal Down syndrome in early pregnancy. Prenat. Diagn. 1993, 13, 633–636. [Google Scholar] [CrossRef]
- Macri, J.; Spencer, K.; Aitken, D.; Garver, K.; Buchanan, P.; Muller, F.; Boue, A. First–trimester free beta (hCG) screening for Down syndrome. Prenat. Diagn. 1993, 13, 557–562. [Google Scholar] [CrossRef]
- Russo, M.; Blakemore, K. A historical and practical review of first trimester aneuploidy screening. Semin. Fetal Neonatal Med. 2014, 19, 183–187. [Google Scholar] [CrossRef]
- Alldred, S.; Takwoingi, Y.; Guo, B.; Pennant, M.; Deeks, J.; Neilson, J.; Alfirevic, Z. First trimester ultrasound tests alone or in combination with first trimester serum tests for Down’s syndrome screening. Cochrane Database Syst. Rev. 2017, 3, CD012600. [Google Scholar] [CrossRef]
- Mackie, F.; Hemming, K.; Allen, S.; Morris, R.; Kilby, M. The accuracy of cell-free fetal DNA-based non-invasive prenatal testing in singleton pregnancies: A systematic review and bivariate meta-analysis. Br. J. Obs. Gynaecol. 2017, 124, 32–46. [Google Scholar] [CrossRef]
- Ogren, L.; Talamantes, F. The Placenta as An Endocrine Organ: Polypeptides. Physiol. Reprod. 1994, 2, 945–975. [Google Scholar]
- Benirschke, K.; Kaufmann, P.; Baergen, R. Pathology of the Human Placenta, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Guibourdenche, J.; Fournier, T.; Malassine, A.; Evain-Brion, D. Development and hormonal functions of the human placenta. Folia. Histochem. Cytobiol. 2009, 47, 35–40. [Google Scholar]
- Olszynko-Gryn, J. The demand for pregnancy testing: The Aschheim-Zondek reaction, diagnostic versatility, and laboratory services in 1930s Britain. Stud. Hist. Philos. Biol. Biomed. Sci. 2013, 47, 233–247. [Google Scholar] [CrossRef]
- Pierce, J.; Parsons, T. Glycoprotein hormones: Structure and function. Ann. Rev. Biochem. 1981, 50, 465–495. [Google Scholar] [CrossRef]
- Ascoli, M.; Segaloff, D. On the structure of the luteinizing hormone/chorionic gonadotropin receptor. Endocr. Rev. 1989, 10, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Casarini, L.; Santi, D.; Brigante, G.; Simoni, M. Two hormones for one receptor: Evolution, biochemistry, actions and pathophysiology of LH and hCG. Endocr. Rev. 2018, 39, 549–592. [Google Scholar] [CrossRef] [PubMed]
- Bahl, O.; Carisen, R.; Bellisario, R.; Swaminathan, N. Human chorionic gonadotrophin: Amino acid sequences of the α and β subunits. J. Biol. Chem. 1975, 250, 5247–5253. [Google Scholar]
- Elliott, M.; Kardana, A.; Lustbader, J.; Cole, L. Carbohydrate and peptide structure of the alpha- and the beta_subunits of human chorionic gonadotropin from normal and aberrant pregnancy and choriocarcinoma. Endocrine 1997, 7, 15–32. [Google Scholar] [CrossRef]
- Talmadge, K.; Boorstein, W.; Fiddes, J. The human genome contains seven genes for the beta-subunit of chorionic gonadotropin but only one gene for the beta-subunit of luteinizing hormone. DNA 1983, 2, 281–289. [Google Scholar] [CrossRef]
- Bo, M.; Boime, I. Identification of the transcriptionally active genes of the chorionic gonadotropin β gene cluster in vivo. J. Biol. Chem. 1992, 267, 3179–3184. [Google Scholar] [CrossRef]
- Camperi, J.; Combes, A.; Guibourdenche, J.; Guillarme, D.; Pichon, V.; Fournier, T.; Vander Heyden, Y.; Mangelings, D.; Delaunay, N. An attempt to characterize the human Chorionic Gonadotropin protein by reversed phase liquid chromatography coupled with high-resolution mass spectrometry at the intact level. J. Pharm. Biomed. Ann. 2018, 161, 35–44. [Google Scholar] [CrossRef]
- Cole, L. hCG, five independent molecules. Clin. Chim. Acta 2012, 413, 48–65. [Google Scholar] [CrossRef]
- Fournier, T.; Guibourdenche, J.; Evain-Brion, D. hCGs: Different sources of production, different glycoforms and functions. Placenta 2015, 36, S60–S65. [Google Scholar] [CrossRef]
- Birken, D.; Maydelman, S.; Gowinowicz, M.; Pound, A.; Liu, Y.; Hartree, A. Isolation and characterization of human pituitary chorionic gonadotropin. Endocrinology 1996, 137, 1405–1411. [Google Scholar] [CrossRef]
- Stenman, U.; Alfthan, H.; Ranta, T.; Vartainen, E.; Jalkanen, J.; Seppata, M. Serum levels of human chorionic gonadotropin in nonpregnant women and men are modulated by gonadotropin-releasing hormone and sex steroids. J. Clin. Endocrinol. Metab. 1987, 64, 730–736. [Google Scholar] [CrossRef]
- Guibourdenche, J.; Handschuh, K.; Tsatsaris, V.; Gerbaud, P.; Leguy, M.C.; Muller, F.; Evain Brion, D.; Fournier, T. Hyperglycosylated hCG is a marker of early human trophoblast invasion. J. Clin. Endocrinol. Metab. 2010, 95, E240–E244. [Google Scholar] [CrossRef]
- Berndt, S.; Blacher, S.; Munaut, C.; Detilleux, J.; Perrier d’Hauterive, S.; Huhtaniemi, I.; Evain-Brion, D.; Noël, A.; Fournier, T.; Foidart, J.M. Hyperglycosylated human chorionic gonadotropin stimulates angiogenesis through TGF-β receptor activation. FASEB J. 2013, 7, 1309–1321. [Google Scholar] [CrossRef]
- Butler, S.; Luttoo, J.; Freire, M.; Abban, T.; Borrelli, P.; Iles, R. Human chorionic gonadotropin (hCG) in the secretome of cultured embryos: Hyperglycosylated hCG and hCG-free beta subunit are potential markers for infertility management and treatment. Reprod. Sci. 2013, 220, 1038–1045. [Google Scholar] [CrossRef]
- Lopata, A.; Oliva, K.; Stanton, P.; Robertson, D. Analysis of chorionic gonadotropin-secreted by cultured human blastocysts. Mol. Hum. Reprod. 1997, 3, 517–521. [Google Scholar] [CrossRef]
- Muyan, M.; Boime, I. Secretion of chorionic gonadotropin from human trophoblasts. Placenta 1997, 18, 237–241. [Google Scholar] [CrossRef]
- Handschuh, K.; Guibourdenche, J.; Tsatsaris, V.; Guesnon, M.; Laurendeau, I.; Evain-Brion, D.; Fournier, T. Human chorionic gonadotropin expression in human trophoblasts from early placenta: Comparative study between villous and extravillous trophoblastic cells. Placenta 2007, 28, 175–184. [Google Scholar] [CrossRef]
- Merz, W. Biosynthesis of human chorionic gonadotropin: A review. Eur. J. Endocrinol. 1996, 135, 269–284. [Google Scholar] [CrossRef]
- Gohar, J.; Mazor, M.; Leiberman, J. GnRH in pregnancy. Arch. Gynecol. Obs. 1996, 259, 1–6. [Google Scholar] [CrossRef]
- Licht, P.; Cao, H.; Lei, Z.; Rao, C.; Merz, W. Novel self-regulation of human chorionic gonadotropin biosynthesis in term pregnancy human placenta. Endocrinology 1993, 133, 3014–3025. [Google Scholar] [CrossRef]
- Chard, T.; Iles, R.; Wathen, N. Why is there a peak of human chorionic gonadotrophin (hCG) in early pregnancy? Hum. Reprod. 1995, 10, 1837–1840. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Rao, C. Gonadotropin receptors in human fetoplacental unit: Implications for HCG as an intracrine, paracrine and endocrine regulator of human fetoplacental function. Troph. Res. 1992, 6, 213–224. [Google Scholar] [CrossRef]
- Cole, L. hCG physiology. Placenta 2013, 34, 1257. [Google Scholar] [CrossRef] [PubMed]
- Birken, S.; Kovalevskaya, G.; O’Connor, J. Metabolism of hCG and hLH to multiple urinary forms. Mol. Cell. Endocrinol. 1996, 125, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Mc Chesney, R.; Wilcox, A.; O’Connor, J.; Weinberg, C.; Baird, D.; Schlatterer, J.; Mc Connaughey, D.; Birken, S.; Canfield, R. Intact hCG, free hCG beta-subunit and hCG betacore fragment: Longitudinal patterns in urine during early pregnancy. Hum. Reprod. 2005, 20, 928–935. [Google Scholar] [CrossRef]
- Korhonen, J.; Alfthan, H.; Ylöstalo, P.; Vedhuis, J.; Stenman, U. Disappearance of human chorionic gonadotropin and its alpha- and beta-subunits after term pregnancy. Clin. Chem. 1997, 43, 2155–2163. [Google Scholar] [CrossRef]
- Nwabuobi, C.; Arlier, S.; Schatz, F.; Guzeloglu-Kayisli, O.; Lockwood, C.; Kayisli, U.A. hCG: Biological functions and clinical applications. Int. J. Mol. Sci. 2017, 18, 2037. [Google Scholar] [CrossRef]
- Perrier d’Hauterive, S.; Close, R.; Gridelet, V.; Mawet, M.; Nisolle, M.; Geenen, V. Human Chorionic Gonadotropin and Early Embryogenesis: Review. Int. J. Mol. Sci. 2022, 23, 1380. [Google Scholar] [CrossRef]
- Tulchinsky, D.; Hobel, C. Plasma human chorionic gonadotropin, estrone, estradiol, estriol, progesterone, and 17 alpha-hydroxyprogesterone in human pregnancy. 3. Early normal pregnancy. Am. J. Obs. Gynecol. 1973, 117, 884–893. [Google Scholar] [CrossRef]
- Casarini, L.; Simoni, M. Recent advances in understanding gonadotropin signaling. Fac. Rev. 2021, 10, 41. [Google Scholar] [CrossRef]
- Troppmann, B.; Kleinau, G.; Krause, G.; Gromoll, J. Structural and functional plasticity of the luteinizing hormone/choriogonadotrophin receptor. Hum. Reprod. Update 2013, 19, 583–602. [Google Scholar] [CrossRef]
- Glinoer, D.; de Nayer, P.; Bourdoux, P.; Lemone, M.; Robyn, C.; van Steirteghem, A.; Kinthaert, J.; Lejeune, B. Regulation of maternal thyroid during pregnancy. J. Clin. Endocrinol. Metab. 1990, 71, 276–287. [Google Scholar] [CrossRef]
- Moleti, M.; Trimarchi, F.; Vermiglio, F. Thyroid physiology in pregnancy. Endocr. Pract. 2014, 20, 589–596. [Google Scholar] [CrossRef]
- Zigmunt, M.; Herr, F.; Keller-Schoenwetter, S.; Kunzi-Rapp, K.; Munstedt, K.; Rao, C.V.; Lang, U.; Preissner, K. Characterization of human chorionic gonadotropin as a novel angiogenic factor. J. Clin. Endocrino. Metab. 2002, 87, 5290–5296. [Google Scholar] [CrossRef]
- Cole, L. Hyperglycosylated hCG. Placenta 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Lee, C.; Chiu, P.; Hautala, L.; Salo, T.; Yeung, W.; Stenman, U.; Koistinen, H. Human chorionic gonadotropin and its free β-subunit stimulate trophoblast invasion independent of LH/hCG receptor. Mol. Cell. Endocrinol. 2013, 375, 43–52. [Google Scholar] [CrossRef]
- Tsampalas, M.; Gridelet, V.; Berndt, S.; Foidart, J.; Geenen, V.; Perrier d’Hauterive, S. Human chorionic gonadotropin: A hormone with immunological and angiogenic properties. J. Reprod. Immunol. 2010, 85, 93–98. [Google Scholar] [CrossRef]
- Schumacher, A.; Heinze, K.; Witte, J.; Poloski, E.; Linzke, N.; Woidacki, K.; Zenclussen, A. Human chorionic gonadotropin as a central regulator of pregnancy immune tolerance. J. Immunol. 2013, 190, 2650–2658. [Google Scholar] [CrossRef]
- Rotmensch, S.; Liberati, M.; Kardana, A.; Copel, J.A.; Ben-Rafael, Z.; Cole, L. Nicked free beta-subunit of human chorionic gonadotropin: A potential new marker for Down syndrome screening. Am. J. Obs. Gynecol. 1996, 174, 609–611. [Google Scholar] [CrossRef]
- Cole, L.; Shahabi, S.; Oz, U.; Bahado-Singh, R.; Mahoney, M. Hyperglycosylated human chorionic gonadotropin (invasive trophoblast antigen) immunoassay: A new basis for gestational Down syndrome screening. Clin. Chem. 1999, 45, 2109–2119. [Google Scholar] [CrossRef]
- Eldar-Gevar, T.; Hochberg, A.; deGroot, N.; Weinstein, D. High maternal serum chorionic gonadotropin level in Downs’ syndrome pregnancies is caused by elevation of both subunits messenger ribonucleic acid level in trophoblasts. J. Clin. Endocrinol. Metab. 1995, 80, 3528–3531. [Google Scholar]
- Jauniaux, E.; Bao, S.; Eblen, A.; Li, X.; Lei, Z.M.; Meuris, S.; Rao, C. HCG concentration and receptor gene expression in placental tissue from trisomy 18 and 21. Mol. Hum. Reprod. 2000, 6, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Smallwood, A.; Chambers, A.; Papageorghiou, A.; Loosfelt, H.; Spencer, K.; Campbell, S.; Nicolaides, K. A link between high serum levels of human chorionic gonadotrophin and chorionic expression of its mature functional receptor (LHCGR) in Down’s syndrome pregnancies. Reprod. Biol. Endocrinol. 2005, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Goshen, R. What factors regulate hCG production in Downs ‘syndrome pregnancies? Mol. Hum. Reprod. 1999, 5, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Aldaz-Carroll, L.; Richon, S.; Dangles-Marie, V.; Cocquebert, M.; Fournier, T.; Troalen, F.; Stevens, D.; Guery, B.; Hersant, A.M.; Guibourdenche, J.; et al. Specific detection of type II human chorionic gonadotropin beta subunit produced by trophoblastic and neoplastic cells. Clin. Chim. Acta 2015, 444, 92–100. [Google Scholar] [CrossRef]
- Cuckle, H.; Iles, R.; Chard, T. Urinary beta-core human chorionic gonadotropin: A new approach to Down’s syndrome screening. Prenat. Diagn. 1994, 14, 953–958. [Google Scholar] [CrossRef]
- Cole, L.; Omrani, A.; Cermik, D.; Singh, R.; Mahoney, M. Hyperglycosylated hCG, a potentiel alternative to hCG in Down syndrome screening. Prenat. Diagn. 1998, 18, 926–933. [Google Scholar] [CrossRef]
- Frendo, J.; Guibourdenche, J.; Pidoux, G.; Vidaud, M.; Luton, D.; Giovangrandi, Y.; Porquet, D.; Muller, F.; Evain-Brion, D. Trophoblast production of a weakly bioactive human chorionic gonadotropin in trisomy 21-affected pregnancy. J. Clin. Endocrinol. Metab. 2004, 89, 727–732. [Google Scholar] [CrossRef]
- Pidoux, G.; Gerbaud, P.; Marpeau, O.; Guibourdenche, J.; Ferreira, F.; Badet, J.; Evain-Brion, D.; Frendo, J. Human placental development is impaired by abnormal human chorionic gonadotropin signaling in trisomy 21 pregnancies. Endocrinology 2007, 148, 5403–5413. [Google Scholar] [CrossRef]
- Brun, S.; Leguy, M.; Bruneel, A.; Fournier, T.; Anselem, O.; Guibourdenche, J. hCG in screening for aneuploidy: A possible role for its glycoforms? Placenta 2014, 35, 425–427. [Google Scholar] [CrossRef]
- Chambers, A.; Mills, W.; Mercade, I.; Crovetto, F.; Crispi, F.; Bodi, L.R.; Pugia, M.; Pira, A.; Lasalvia, L.; Banerjee, S.; et al. The utility of circulating LHCGR as a predictor of Down’s syndrome in early pregnancy. BMC Pregnancy Childbirth 2014, 14, 197. [Google Scholar] [CrossRef]
- Lin, T.; Halbert, S.; Spellacy, W. Measurement of pregnancy-associated plasma proteins during gestation. J. Clin. Investig 1974, 54, 576–582. [Google Scholar] [CrossRef]
- Conover, C.A. Key questions and answers about pregnancy-associated plasma protein-A. Trends Endocrinol. Metab. 2012, 23, 242–249. [Google Scholar] [CrossRef]
- Oxvig, C.; Sand, O.; Kristensen, T.; Gleich, G.J.; Sottrup-Jensen, L. Circulating human pregnancy-associated plasma protein-A is disulfide-bridged to the preform of eosinophil major basic protein. J. Biol. Chem. 1993, 268, 12243–12246. [Google Scholar] [CrossRef]
- Oxvig, C.; Sand, O.; Kristensen, T.; Kristensen, L.; Sottrup-Jensen, L. Isolation and characterization of circulating complex between human pregnancy associated plasma protein A and proform of eosinophil major basic protein. Biochem. Biophys. Acta 1994, 1201, 415–423. [Google Scholar] [CrossRef]
- Kristensen, T.; Oxvig, C.; Sand, O.; Moller, N.; Sottrup-Jensen, L. Ammino acid sequence of human pregnancy associated plasma protein-A derived from cloned cDNA. Biochemistry 1994, 33, 1592–1598. [Google Scholar] [CrossRef]
- Farr, M.; Strübe, J.; Geppert, H.; Kocourek, A.; Mahne, M.; Tschesche, H. Pregnancy-associated plasma protein-E (PAPP-E). Biochem. Biophys. Acta 2000, 1493, 356–362. [Google Scholar] [CrossRef]
- Kirkegaard, I.; Uldberg, N.; Oxvig, C. Biology of pregnancy-associated plasma protein-A in relation to prenatal diagnostics: An overview. Act. Obs. Gynecol. 2010, 89, 1118–1125. [Google Scholar] [CrossRef]
- Monget, P.; Oxvig, C. PAPP-A and the IGF system. Ann. Endocrinol. 2016, 77, 90–96. [Google Scholar] [CrossRef]
- Overpris, S.; Skov, L.; Glerup, S.; Pihl, K.; Christiansen, M.; Oxvig, C. Formation of high-molecular-weight angiotensinogen during pregnancy is a result of competing redox reactions with the proform of eosinophil major basic protein. Biochem. J. 2013, 449, 209–217. [Google Scholar] [CrossRef]
- Overgaard, M.T.; Oxvig, C.; Christiansen, M.; Lawrence, J.B.; Conover, C.A.; Gleich, G.J.; Sottrup-Jensen, L.; Haaning, J. Messenger Ribonucleic Acid Levels of Pregnancy-Associated Plasma Protein-A and the Proform of Eosinophil Major Basic Protein: Expression in Human Reproductive and Nonreproductive Tissues. Biol. Reprod. 1999, 61, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.; Bordignon, P.; Bischof, P. Immunohistochemical localization of pregnancy associated plasma protein-A in decidua and trophoblast: Comparison with human chorionic gonadotrophin and fibrin. Placenta 1984, 5, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Halbert, S. Placental localization of human pregnancy-associated plasma proteins. Science 1986, 193, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Bonno, M.; Oxvig, C.; Kephart, G.M.; Wagner, J.M.; Kristensen, T.; Sottrup-Jensen, L.; Gleich, G. Localization of pregnancy associated plasma protein-A and colocalization of pregnancy associated plasma protein-A messenger ribonucleic acid and eosinophil granule major basic protein messenger ribonucleic acid in placenta. La. Investig. 1994, 71, 560–566. [Google Scholar]
- Guibourdenche, J.; Frendo, J.; Pidoux, G.; Bertin, G.; Luton, D.; Muller, F.; Porquet, D.; Evain-Brion, D. Expression of pregnancy-associated plasma protein-A(PAPP-A) during villous trophoblast differentiation in vitro. Placenta 2003, 24, 532–539. [Google Scholar] [CrossRef]
- D’Elia, P.; Marzioni, D.; Castellucci, M.; Locci, C.; Pala, A. Homodimeric pregnancy-associated plasma protein-A in normal human placenta of first and third trimester of pregnancy: Biochemical and morphological observations. Placenta 2012, 33, 942–945. [Google Scholar] [CrossRef]
- Folkersen, J.; Grudzinskas, J.; Hindersson, P.; Teisner, B.; Westergaard, J. Pregnancy associated plasma protein A: Circulating levels during normal pregnancy. Am. J. Obs. Gynecol. 1981, 139, 910–924. [Google Scholar] [CrossRef]
- Metzenbauer, M.; Hafner, E.; Hoefinger, D.; Schuchter, K.; Stangl, G.; Ogris, E.; Philipp, K. Three-dimensional ultrasound measurement of the placental volume in early pregnancy: Method and correlation with biochemical placenta parameters. Placenta 2001, 22, 602–605. [Google Scholar] [CrossRef]
- Sahraravand, M.; Järvelä, I.; Laitinen, P.; Tekay, A.; Ryynänen, M. The secretion of PAPP-A, ADAM 12 and PP13 correlates with the size of the placenta for the first month of pregnancy. Placenta 2011, 32, 999–1003. [Google Scholar] [CrossRef]
- Christiansen, M.; Oxvig, C.; Wagner, J.; Qin, Q.; Nguyen, T.; Overgaard, M.; Larsen, S.; Sottrup-Jensen, L.; Gleich, G.; Nogaard-Dedersen, B. The proform of eosinophil major basic protein: A new maternal serum marker for Down Syndrome. Prenat. Diagn. 1999, 19, 905–910. [Google Scholar] [CrossRef]
- Bersinger, N.; Xin, W.; Birkhäuser, M. Glycosylation of pregnancy associated plasma protein-A (PAPP-A) in normal and trisomic pregnancies: Studies with lectins. Troph. Res. 1999, 13, 493–501. [Google Scholar] [CrossRef]
- Oxvig, C.; Conover, C. The Stanniocalcin-PAPP-A-IGFBP-IGF Axis. J. Clin. Endocrinol. Metab. 2023, 31, dgad053. [Google Scholar] [CrossRef]
- Judge, R.; Sridar, J.; Tunyasuvunakool, K.; Jain, R.; Wang, J.; Ouch, C.; Xu, J.; Mafi, A.; Nile, A.; Remarcik, C.; et al. Structure of the PAPP-ABP5 complex reveals mechanism of substrate recognition. Nat. Commun. 2022, 13, 5500. [Google Scholar] [CrossRef]
- Lawrence, J.; Oxvig, C.; Overgaard, M.; Sottrup-Jensen, L.; Gleich, G.; Hays, L.; Yates, J.; Conover, C. The insulin-like growth factor (IGF)-dependent IGF binding protein-4 protease secreted by human fibroblasts is pregnancy associated plasma protein -A. Proc. Natl. Acad. Sci. USA 1999, 96, 3149–3153. [Google Scholar] [CrossRef]
- Yan, X.; Baxter, R.; Firth, S. Involvement of pregnancy-associated plasma protein-A2 in insulin-like growth factor (IGF) binding protein-5 proteolysis during pregnancy: A potential mechanism for increasing IGF bioavailability. J. Clin. Endocrinol. Metab. 2010, 95, 1412–1420. [Google Scholar] [CrossRef]
- Giuduce, L.; Conover, C.; Bale, L.; Faessen, G.; Ilg, K.; Sun, I.; Imani, B.; Suen, L.; Irwin, J.; Christiansen, M.; et al. Identification and regulation of the IGFBP-4 protease and its physiological inhibitor in human trophoblasts and endometrial stroma: Evidence for paracrin regulation of IGF-II bioavailability in the placental bed during human implantation. J. Clin. Endocrinol. Metab. 2002, 87, 2359–2366. [Google Scholar] [CrossRef]
- Rojas-Rodriguez, R.; Ziegler, R.; DeSouza, T.; Majid, S.; Madore, A.; Amir, N.; Pace, V.; Nachreiner, D.; Alfego, D.; Mathew, J.; et al. Papp-a mediated adipose tissue remodeling mitigates insulin resistance and protects against gestational diabetes in mice and humans. Sci. Transl. Med. 2020, 12, eaay4145. [Google Scholar] [CrossRef]
- Overgaard, M.; Haaning, J.; Boldt, H.; Olsen, I.; Laursen, L.; Christiansen, M.; Gleich, G.; Sottrup-Jensen, L.; Conover, C.; Oxvig, C. Expression of recombinant human pregnancy associated plasma protein A and identification of the proform eosinophil major basic protein as its physiological inhibitor. J. Biol. Chem. 2000, 275, 31128–311433. [Google Scholar] [CrossRef]
- Weyer, K.; Glerup, S. Placental regulation of peptide hormone and growth factor activity by proMB. Biol. Reprod. 2011, 84, 1077–1086. [Google Scholar] [CrossRef]
- Bishop, A.; Cartwright, J.; Whitley, G. Stanniocalcin-1 in the female reproductive system and pregnancy. Hum Reprod Update 2021, 27, 1098–1114. [Google Scholar] [CrossRef]
- Kobber, S.; Gajhede, M.; Mirza, O.A.; Kloverpris, S.; Kjaer, T.; Mikkelsen, J.; Boesen, T.; Oxvig, C. Structure of the proteolytic enzyme PAPP-A with the endogenous inhibitor stanniocalcin-2 reveals its inhibitory mechanism. Nat. Comm. 2022, 13, 6084. [Google Scholar] [CrossRef] [PubMed]
- Handschuh, K.; Guibourdenche, J.; Guesnon, M.; Laurendeau, I.; Evain-Brion, D.; Fournier, T. Modulation of PAPP-A expression by PPARγ in human first trimester trophoblast. Placenta 2006, 27, S127–S134. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Qin, H.; Zhao, Y.; Wang, X.; Yan, Q. Pregnancy-associated plasma protein A up-regulated by progesterone promotes adhesion and proliferation of trophoblastic cells. Int. J. Clin. Exp. Pathol. 2014, 7, 1427–1437. [Google Scholar] [PubMed]
- Haddow, J.; Palomaki, G.; Knight, G.; Williams, J.; Miller, W.; Johnson, A. Screening of maternal serum for fetal Down’s syndrome in the first trimester. N. Engl. J. Med. 1998, 338, 955–961. [Google Scholar] [CrossRef]
- Brizot, M.; Hyette, J.; Mckie, A.; Bersinger, N.; Farzaneh, F.; Nicolaides, K. Gene expression of human pregnancy-associated plasma protein-A in placenta from trisomic pregnancies. Placenta 1996, 17, 33–36. [Google Scholar] [CrossRef]
- Tul, N.; Spencer, K.; Noble, P.; Chan, C.; Nicolaides, K. Screening for trisomy 18 by fetal nuchal translucency and maternal serum free beta-hCG and PAPP-A at 10–14 weeks of gestation. Prenat. Diagn. 1999, 19, 1035–1042. [Google Scholar] [CrossRef]
- Aitken, D.; Ireland, M.; Berry, E.; Crossley, J.; Macri, J.; Burn, J.; Connor, J. Second trimester pregnancy associated plasma protein-A levels are reduced in Cornelia de Lange syndrome pregnancies. Prenat. Diagn. 1999, 19, 706–710. [Google Scholar] [CrossRef]
- Westergaard, J.; Sinosich, M.; Bugge, M.; Madsen, L.; Teisner, B.; Grudzinskas, J. Pregnancy-associated plasma protein A in the prediction of early pregnancy failure. Am. J. Obs. Gynecol. 1983, 145, 67–69. [Google Scholar] [CrossRef]
- Kaijomaa, M.; Rahkonen, L.; Ulander, V.; Hämäläinen, E.; Alfthan, H.; Markkanen, H.; Heinonen, S.; Stefanovic, V. Low maternal pregnancy-associated plasma protein A during the first trimester of pregnancy and pregnancy outcomes. Int. J. Gynaecol. Obs. 2017, 136, 76–82. [Google Scholar] [CrossRef]
- Petraglia, F.; Sawchenko, P.; Lim, A.T.; Rivier, J.; Vale, W. Localization, secretion, and action of inhibin in human placenta. Science 1987, 237, 187–189. [Google Scholar] [CrossRef]
- Petraglia, F. Inhibin, activin and follistatin in the human placenta--a new family of regulatory proteins. Placenta 1997, 18, 3–8. [Google Scholar] [CrossRef]
- Adu-Gyamfi, E.; Ding, Y.; Wang, Y. Regulation of placentation by the transforming growth factor beta superfamily. Biol. Reprod. 2020, 102, 18–26. [Google Scholar] [CrossRef]
- Debieve, F.; Moiset, A.; Thomas, K.; Pampfer, S.; Hubinont, C. Vascular endothelial growth factor and placenta growth factor concentrations in Down’s syndrome and control pregnancies. Mol. Hum. Reprod. 2001, 8, 765–770. [Google Scholar] [CrossRef]
- Wald, N.; Bestwick, J.; George, L.; Huttly, W. Antenatal screening for Down syndrome using serum placental growth factor with the combined, quadruple, serum integrated and integrated tests. PLoS ONE 2012, 7, e46955. [Google Scholar] [CrossRef]
- Van Lith, J.; Pratt, J.; Beekhuis, J.; Mantingh, A. Second trimester maternal serum immunoreactive inhibin as a marker for fetal Down’s syndrome. Prenat. Diagn. 1992, 12, 801–806. [Google Scholar] [CrossRef]
- Makanji, Y.; Zhu, J.; Mishra, R.; Holmquist, C.; Wong, W.; Schwartz, N.; Mayo, K.; Woodruff, T. Inhibin at 90: From discovery to clinical application, a historical review. Endocr. Rev. 2014, 35, 747–794. [Google Scholar] [CrossRef]
- Namwanje, M.; Brown, C. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a021881. [Google Scholar] [CrossRef]
- Walton, K.; Makanji, Y.; Robertson, D.; Harrison, C. The synthesis and secretion of inhibins. Vitam. Horm. 2011, 85, 149–184. [Google Scholar]
- Barton, D.; Yang-Feng, T.; Mason, A.; Seeburg, P.; Francke, U. Mapping of genes for inhibin subunits alpha, beta A, and beta B on human and mouse chromosomes and studies of jsd mice. Genomics 1989, 5, 91–99. [Google Scholar] [CrossRef]
- Makanji, Y.; Harrison, C.A.; Stanton, P.G.; Krishna, R.; Robertson, D. Inhibin A and B in vitro bioactivities are modified by their degree of glycosylation and their affinities to betaglycan. Endocrinology 2007, 148, 2309–2316. [Google Scholar] [CrossRef]
- Cook, R.; Thompson, T.; Jardetzky, T.; Woodruff, T. Molecular biology of inhibin action. Semin. Reprod. Med. 2004, 22, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F. Activin A in mammalian physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Kolsky, M.; Manuelpillai, U.; Waldron, K.; Dole, A.; Wallace, E. The distribution of activin and activin receptors in gestational tissues across human pregnancy and during labour. Placenta 2002, 23, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Caniggia, I.; Lye, S.; Cross, J. Activin is a local regulator of human cytotrophoblast cell differentiation. Endocrinology 1997, 138, 3976–3986. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Keelan, J.; France, J. Activin-A stimulates, while transforming growth factor beta 1 inhibits, chorionic gonadotrophin production and aromatase activity in cultured human placental trophoblasts. Placenta 1996, 17, 603–610. [Google Scholar] [CrossRef]
- Li, Y.; Klausen, C.; Zhu, H.; Leung, P.C. Activin A Increases Human Trophoblast Invasion by Inducing SNAIL-Mediated MMP2 Up-Regulation Through ALK4. J. Clin. Endocrinol. Metab. 2015, 100, E1415–E1427. [Google Scholar] [CrossRef]
- Watt, H.; Wald, N.; Huttly, W. The pattern of maternal serum inhibin-A concentrations in the second trimester of pregnancy. Prenat. Diagn. 1998, 18, 846–848. [Google Scholar] [CrossRef]
- Birdsall, M.; Ledger, W.; Groome, N.; Abdalla, H.; Muttukrishna, S. Inhibin A and activin A in the first trimester of human pregnancy. J. Clin. Endocrinol. Metab. 1997, 82, 1557–1560. [Google Scholar] [CrossRef]
- O’Connor, A.; McFarlane, J.; Hayward, S.; Yohkaichiya, T.; Groome, N.; de Kretser, D. Serum activin A and follistatin concentrations during human pregnancy: A cross-sectional and longitudinal study. Hum. Reprod. 1999, 14, 827–832. [Google Scholar] [CrossRef]
- Fowler, P.; Evans, L.; Groome, N.; Templeton, A.; Knight, P. A longitudinal study of maternal serum inhibin-A, inhibin-B, activin-A, activin-AB, pro-alphaC and follistatin during pregnancy. Hum. Reprod. 1998, 13, 3530–3536. [Google Scholar] [CrossRef]
- Lambert-Messerlian, G.; Pinar, H.; Laprade, E.; Tantravahi, U.; Schneyer, A.; Canick, J. Inhibins and activins in human fetal abnormalities. Mol. Cell. Endocrinol. 2004, 225, 101–108. [Google Scholar] [CrossRef]
- Muttukrishna, S.; Knight, P.; Groome, N.; Redman, C.; Ledger, W. Activin A and inhibin A as possible endocrine markers for pre-eclampsia. Lancet 1997, 349, 1285–1288. [Google Scholar] [CrossRef]
- Wald, N.; Densem, J.; George, L.; Muttukrishna, S.; Knight, P. Prenatal screening for Down’s syndrome using inibin-A as a serum marker. Prenat. Diagn. 1996, 16, 143–153. [Google Scholar] [CrossRef]
- Dalgliesh, G.; Aitken, D.; Lyall, F.; Howatson, A.; Connor, J. Placental and maternal serum inhibin-A and activin-A levels in Down’s syndrome pregnancies. Placenta 2001, 22, 227–234. [Google Scholar] [CrossRef]
- Thirunavukarasu, P.; Lambert-Messerlian, G.; Robertson, D.; Dawson, G.; Canick, J.; Wallace, E. Molecular weight forms of inhibin A, inhibin B and pro-alphaC in maternal serum, amniotic fluid and placental extracts of normal and Down syndrome pregnancies. Prenat. Diagn. 2002, 22, 1086–1092. [Google Scholar] [CrossRef]
- Kipp, J.; Lambert-Messerlian, G.; Eklund, E.; Rodriguez, G.; Demczuk, M.; Gundogan, F. Expression of transcription factors controlling alpha inhibin gene expression in placental tissues from pregnancies affected by fetal Down syndrome. Prenat. Diagn. 2012, 32, 302–305. [Google Scholar] [CrossRef]
- Gerbaud, P.; Pidoux, G.; Guibourdenche, J.; Pathirage, N.; Costa, J.; Badet, J.; Frendo, J.; Murthi, P.; Evain-Brion, D. Mesenchymal activin A reverses abnormal trisomy 21 trophoblast fusion and differentiation. Endocrinology 2011, 152, 5017–5028. [Google Scholar] [CrossRef]
- Deutsch, H. Chemistry and biology of alpha-fetoprotein. Adv. Cancer Res. 1991, 56, 253–312. [Google Scholar]
- Terentiev, A.; Moldogazieva, N. Alpha-fetoprotein: A renaissance. Tumour Biol. 2013, 34, 2075–2091. [Google Scholar] [CrossRef]
- Law, S.; Dugaiczyk, A. Homology between the primary structure of alpha-fetoprotein, deduced from a complete cDNA sequence, and serum albumin. Nature 1981, 291, 201–205. [Google Scholar] [CrossRef]
- Mizejewski, G. Alpha-fetoprotein structure and function: Relevance to isoforms, epitopes, and conformational variants. Exp. Biol. Med. 2001, 226, 377–408. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, T.; Sakai, M.; Wegmann, T.; Tamaoki, T. Primary structures of human alpha-fetoprotein and its mRNA. Proc. Natl. Acad. Sci. USA 1983, 80, 4604–4608. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Naumova, A.; Liebhaber, S.; Cooke, N. Physical and meiotic mapping of the region of human chromosome 4q11-q13 encompassing the vitamin D binding protein DBP/Gc-globulin and albumin multigene cluster. Genome Res. 1999, 9, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Katoh, H.; Yamashita, F.; Tanaka, M.; Tanikawa, K.; Taketa, K.; Satomura, S.; Matsuura, S. Comparison of carbohydrate structures of serum -fetoprotein by sequential glycosidase digestion and lectin affinity electrophoresis. Clin. Chim. Acta 1996, 254, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, D.; Boesman, M. Sites of serum alpha-fetoprotein synthesis in the human and in the rat. Clin. Investig. 1967, 46, 1010–1016. [Google Scholar] [CrossRef]
- Lafuste, P.; Robert, B.; Mondon, F.; Danan, J.L.; Rossi, B.; Duc-Goiran, P.; Mignot, T.M.; Nunez, E.A.; Benassayag, C.; Ferré, F. Alpha-fetoprotein gene expression in early and full-term human trophoblast. Placenta 2002, 23, 600–612. [Google Scholar] [CrossRef]
- Duc-Goiran, P.; Mignot, T.; Robert, B.; Machavoine, F.; Mondon, F.; Hagneré, A.M.; Vacher-Lavenu, M.; Danan, J.; Vaiman, D.; Benassayag, C.; et al. Expression and localization of alpha-fetoprotein mRNA and protein in human early villous trophoblasts. Placenta 2006, 27, 812–821. [Google Scholar] [CrossRef]
- Gitlin, D. Sites of alpha-fetoprotein synthesis. N. Engl. J. Med. 1971, 285, 1436–1437. [Google Scholar]
- Ikonen, R.; Lindgren, J.; Niemi, E.; Sorto, A.; Seppälä, M.; Ruoslahti, E. Alpha fetoprotein levels in neonatal hyperbilirubinaemia. Acta Paediatr. Scand. 1980, 69, 59–63. [Google Scholar] [CrossRef]
- Gordon, Y.; Ratky, S.; Leighton, P.; Kitau, M.; Chard, T. Amniotic fluid levels of fibrin(ogen) degradation fragment E and alpha-fetoprotein in normal pregnancy and with fetal neural tube defect. Br. J. Obs. Gynaecol. 1976, 83, 771–774. [Google Scholar] [CrossRef]
- Los, F.; De Bruijn, H.; van Beek Calkoen-Carpay, T.; Huisjes, H. AFP transport across the fetal membranes in the human. Prenat. Diagn. 1985, 5, 277–281. [Google Scholar] [CrossRef]
- Brownbill, P.; Edwards, D.; Jones, C.; Mahendran, D.; Owen, D.; Sibley, C.; Johnson, R.; Swanson, P.; Nelson, D. Mechanisms of AFP transfer in the perfused human placental cotyledon from uncomplicated pregnancy. J. Clin. Investig. 1995, 96, 2220–2226. [Google Scholar] [CrossRef]
- Malek, A.; Sager, R.; Schneider, H. Transport of protein across the human placenta. Am. J. Reprod. Immunol. 1998, 40, 347–351. [Google Scholar] [CrossRef]
- Newby, D.; Dalgliesh, G.; Lyall, F.; Aitken, D. Alphafetoprotein and alphafetoprotein receptor expression in the normal human placenta at term. Placenta 2005, 26, 190–200. [Google Scholar] [CrossRef]
- Chen, H.; Egan, J.; Chiu, J. Regulation and activities of alpha-fetoprotein. Clin. Rev. Eukar. Gene Expr. 1997, 7, 11–41. [Google Scholar] [CrossRef]
- Tsukada, Y.; Hirai, H. Alpha-Fetoprotein and albumin synthesis during the cell cycle. Ann. N. Y Acad. Sci. 1975, 259, 37–44. [Google Scholar] [CrossRef]
- Kajiyama, Y.; Tian, J.; Locker, J. Regulation of alpha-fetoprotein expression by Nkx2.8. Mol. Cell. Biol. 2002, 22, 6122–6130. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, Y.; Tian, J.; Locker, J. Characterization of distant enhancers and promoters in the albumin-alpha-fetoprotein locus during active and silenced expression. J. Biol. Chem. 2006, 281, 30122–30131. [Google Scholar] [CrossRef] [PubMed]
- Mizejewski, G. Biological roles of alpha-fetoprotein during pregnancy and perinatal development. Exp. Biol. Med. 2004, 229, 439–463. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Galgano, M.; Mutinati, M.; Sciorsci, R. Alpha-fetoprotein in animal reproduction. Res. Vet. Sci. 2019, 123, 281–285. [Google Scholar] [CrossRef]
- Mizejewski, G. Physiology of alpha-fetoprotein as a biomarker for perinatal distress: Relevance to adverse pregnancy outcome. Exp. Biol. Med. 2007, 232, 993–1004. [Google Scholar] [CrossRef]
- Toder, V.; Blank, M.; Gold-Gefter, L.; Nebel, L. The effect of alpha-fetoprotein on the growth of placental cells in vitro. Placenta 1983, 4, 79–86. [Google Scholar] [CrossRef]
- Yachnin, S.; Getz, G.; Lusk, L.; Hsu, R. Lipid interactions with human alpha-fetoprotein (AFP). A study of the role of such interactions in the ability of human AFP to suppress lymphocyte transformation. Oncodev Biol. Med. 1980, 1, 273–285. [Google Scholar]
- Vranckx, R.; Savu, L.; Maya, M.; Nunez, E. Alpha-feotoprotein and transcortin behave as acute phase reactants in the maternal and fetal compartments of the inflammatory pregnant mouse. Endocrinology 1987, 120, 1782–1789. [Google Scholar] [CrossRef]
- Laderoute, M.; Pilarski, L. The inhibition of apoptosis by alpha-fetoprotein (AFP) and the role of AFP receptors in anti-cellular senescence. Anticancer Res. 1994, 14, 2429–2438. [Google Scholar]
- Uriel, J.; Bouillon, D.; Aussel, C.; Dupiers, M. Alpha-fetoprotein: The major high-affinity estrogen binder in rat uterine cytosols. Proc. Natl. Acad. Sci. USA 1976, 73, 1452145–1452146. [Google Scholar] [CrossRef]
- Hsia, J.; Er, S.; Tan, C.; Estes, T.; Ruoslahti, E. Alpha-fetoprotein binding specificity for arachidonate, bilirubin, docosahexaenoate, and palmitate. A spin label study. J. Biol. Chem. 1980, 255, 4224–4227. [Google Scholar] [CrossRef]
- Wu, J.; Monir-Vaghefi, S.; Clayton, F. Human alpha-fetoprotein and albumin: Differences in zinc binding. Clin. Physiol. Biochem. 1987, 5, 85–94. [Google Scholar]
- Lorber, J.; Stewart, C.; Ward, A. Alpha-fetoprotein in antenatal diagnosis of anencephaly and spina bifida. Lancet 1973, 1, 1187. [Google Scholar] [CrossRef]
- Leighton, P.; Kitau, M.; Chard, T.; Gordon, Y.; Leek, A. Levels of alpha-fetoprotein in maternal blood as a screening test for fetal neural-tube defect. Lancet 1975, 2, 1012–1015. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Lu, S.; Huang, J.; Zhang, L.; Hu, W. The diagnostic accuracy of maternal serum alpha-fetoprotein variants (AFP-L2 and AFP-L3) in predicting fetal open neural tube defects and abdominal wall defects. Clin. Chim. Acta 2020, 507, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Alldred, S.; Deeks, J.; Guo, B.; Neilson, J.; Alfirevic, Z. Second trimester serum tests for Down’s Syndrome screening. Cochrane Database Syst. Rev. 2012, 6, CD009925. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Dennis, A.; Meschino, W.S.; Rashid, S.; Mak-Tam, E.; Cuckle, H. First trimester screening for Down syndrome using nuchal translucency, maternal serum pregnancy-associated plasma protein A, free-β human chorionic gonadotrophin, placental growth factor, and α-fetoprotein. Prenat. Diagn. 2015, 35, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Gulbis, B.; Gervy, C.; Jauniaux, E. Amniotic fluid biochemistry in second-trimester trisomic pregnancies: Relationships to fetal organ maturation and dysfunction. Early Hum. Dev. 1998, 52, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Kronquist, K.; Dreazen, E.; Keener, S.; Nicholas, T.; Crandall, B. Reduced fetal hepatic alpha-fetoprotein levels in down’s syndrome. Prenat. Diagn. 1990, 10, 739–751. [Google Scholar] [CrossRef]
- Brizot, M.; Mckie, A.; von Kaisenberg, C.; Farzaneh, F.; Nicolaides, K. Fetal hepatic alpha-fetoprotein mRNA expression in fetuses with trisomy 21 and 18 at 12–15 weeks gestation. Early Hum. Dev. 1996, 44, 155–159. [Google Scholar] [CrossRef]
- Los, F.; Janse, H.; Brandenburg, H.; de Vrij, R.; de Bruijn Henk, W. Concanavalin A variants of alpha-fetoprotein in first trimester fetuses with trisomy 21 and with normal karyotypes. Gynecol. Obs. Investig. 1995, 9, 149–152. [Google Scholar] [CrossRef]
- Yamamoto, R.; Azuma, M.; Kishida, T.; Yamada, H.; Satomura, S.; Fujimoto, S. Total alpha-fetoprotein and Lens culinaris agglutinin-reactive alpha-fetoprotein in fetal chromosomal abnormalities. BJOG 2001, 108, 1154–1158. [Google Scholar]
- Chen, Y.; Chen, Y.; Ning, W.; Zhang, W.; Li, L.; Wang, X.; Yin, Y.; Zhang, H. Diagnostic value of maternal alpha-fetoprotein variants in second-trimester biochemical screening for trisomy 21 and 18. Sci. Rep. 2022, 12, 13605. [Google Scholar] [CrossRef]
- Newby, D.; Aitken, D.; Crossley, J.; Howatson, A.; Macri, J.; Connor, J. Biochemical markers of trisomy 21 and the pathophysiology of Down’s syndrome pregnancies. Prenat. Diagn. 1997, 17, 941–951. [Google Scholar] [CrossRef]
- Diczfalusy, E.; Magnusson, A. Estriol in blood. J. Clin. Endocrinol. Metab. 1960, 20, 1633–1634. [Google Scholar] [CrossRef]
- Siiteri, P.; MacDonald, P. Placental estrogen biosynthesis during human pregnancy. J. Clin. Endocrinol. Metab. 1966, 26, 751–761. [Google Scholar] [CrossRef]
- Villee, C. Placenta and fetal tissues: A biphasic system for the synthesis of steroids. Am. J. Obs. Gynecol. 1969, 104, 406–415. [Google Scholar] [CrossRef]
- Albrecht, E.; Pepe, G. Placental steroid hormone biosynthesis in primate pregnancy. Endocr. Rev. 1990, 11, 124–150. [Google Scholar] [CrossRef]
- Pasqualini, J. Enzymes involved in the formation and transformation of steroid hormones in the fetal and placental compartments. J. Steroid Biochem. Mol. Biol. 2005, 97, 401–415. [Google Scholar] [CrossRef]
- Morel, Y.; Roucher, F.; Plotton, I.; Goursaud, C.; Tardy, V.; Mallet, D. Evolution of steroids during pregnancy: Maternal, placental and fetal synthesis. Ann. Endocrinol. 2016, 77, 82–89. [Google Scholar] [CrossRef]
- Warren, J.; French, A. Distribution of steroid sulfatase in human tissues. J. Clin. Endocrinol. Metab. 1965, 25, 278–282. [Google Scholar] [CrossRef]
- Harada, N.; Yoshimura, N.; Honda, S. Unique regulation of expression of human aromatase in the placenta. J. Steroid Biochem. Mol. Biol. 2003, 86, 327–334. [Google Scholar] [CrossRef]
- Chatuphonprasert, W.; Jarukamjorn, K.; Ellinger, I. Physiology and Pathophysiology of Steroid Biosynthesis, Transport and Metabolism in the Human Placenta. Front. Pharm. 2018, 9, 1027. [Google Scholar] [CrossRef]
- Mendelson, C.; Kamat, A. Mechanisms in the regulation of aromatase in developing ovary and placenta. J. Steroid Biochem. Mol. Biol. 2007, 106, 62–70. [Google Scholar] [CrossRef]
- De Hertogh, R.; Thomas, K.; Bietlot, Y.; Vanderheyden, I.; Ferin, J. Plasma levels of unconjugated estrone, estradiol and estriol and of HCS throughout pregnancy in normal women. J. Clin. Endocrinol. Metab. 1975, 40, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.; Ket, J.; Caanen, M.; Lambalk, C. Reproductive hormone concentrations in pregnancy and neonates: A systematic review. Reprod. Biomed. Online 2013, 27, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Moutsatsou, V.; Oakey, R. Oestriol binding to plasma proteins. J. Steroid Biochem. 1988, 29, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.; Korach, K. Estrogen Receptors: New Directions in the New Millennium. Endocr. Rev. 2018, 39, 664–675. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Lee, Y.; Joo, J.; An, B. Interaction of steroid receptor coactivators and estrogen receptors in the human placenta. J. Mol. Endocrinol. 2016, 56, 239–247. [Google Scholar] [CrossRef]
- Charles, S.; Julian, C.; Vargas, E.; Moore, L. Higher estrogen levels during pregnancy in Andean than European residents of high altitude suggest differences in aromatase activity. J. Clin. Endocrinol. Metab. 2014, 99, 2908–2916. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Lefevre, G.; Alfaidy, N.; Nahed, R.; Vincent, J.; Oudinet, J.; Pianos, A.; Cambourg, A.; Rozenberg, P.; et al. Abnormal steroidogenesis and aromatase activity in preeclampsia. Placenta 2018, 69, 40–49. [Google Scholar] [CrossRef]
- Mucci, L.; Lagiou, P.; Tamimi, R.; Hsieh, C.; Adami, H.; Trichopoulos, D. Pregnancy estriol, estradiol, progesterone and prolactin in relation to birth weight and other birth size variables (United States). Cancer Causes Control. 2003, 14, 311–318. [Google Scholar] [CrossRef]
- Falah, N.; Torday, J.; Quinney, S.K.; Haas, D. Estriol review: Clinical applications and potential biomedical importance. Clin. Res. Trials 2015, 1, 29–33. [Google Scholar] [CrossRef]
- Lupo, V.; Miodovnik, M.; Hertzberg, V. Daily variation of serum unconjugated estriol and estetrol in normal pregnancy. Am. J. Perinatol. 1985, 2, 330–337. [Google Scholar] [CrossRef]
- Huang, X.; Spink, D.; Schneider, E.; Ling, H.; Rai, A.; Rosano, T.; Chen, B.; Cao, Z. Measurement of unconjugated estriol in serum by liquid chromatography-tandem mass spectrometry and assessment of the accuracy of chemiluminescent immunoassays. Clin. Chem. 2014, 60, 260–268. [Google Scholar] [CrossRef]
- Crandall, B.; Hanson, F.; Keener, S.; Matsumoto, M.; Miller, W. Maternal serum screening for alpha-fetoprotein, unconjugated estriol, and human chorionic gonadotropin between 11 and 15 weeks of pregnancy to detect fetal chromosome abnormalities. Am. J. Obs. Gynecol. 1993, 168, 1864–1867. [Google Scholar] [CrossRef]
- Spencer, K.; Muller, F.; Aitken, D. Biochemical markers of trisomy 21 in amniotic fluid. Prenat. Diagn. 1997, 17, 31–37. [Google Scholar] [CrossRef]
- Newby, D.; Aitken, D.; Howatson, A.; Connor, J. Placental synthesis of oestriol in Down’s syndrome pregnancies. Placenta 2000, 21, 263–267. [Google Scholar] [CrossRef]
- Landt, J.; Ball, S.; Holland, A.; Hon, J.; Owen, A.; Treppner, P.; Herbert, J. Age-related changes in plasma dehydroepiandrosterone levels in adults with Down’s syndrome and the risk of dementia. J. Neuroendocr. 2011, 23, 450–455. [Google Scholar] [CrossRef]
- Pang, Y.; Wang, C.; Tang, J.; Zhu, J. Clinical application of noninvasive prenatal testing in the detection of fetal chromosomal diseases. Mol. Cytogenet. 2021, 14, 31. [Google Scholar] [CrossRef]
- Vossaert, L.; Chakchouk, I.; Zemet, R.; Van den Veyver, I. Overview and recent developments in cell-based non invasive prenatal testing. Prenat. Diagn. 2021, 41, 1202–1214. [Google Scholar] [CrossRef]
- Walknowska, J.; Conte, F.; Grumbach, M. Practical and theoretical implications of fetal-maternal lymphocyte transfer. Lancet 1969, 1, 1119–1122. [Google Scholar] [CrossRef]
- Herzenberg, L.; Bianchi, D.; Schröder, J.; Cann, H.; Iverson, G. Fetal cells in the blood of pregnant women: Detection and enrichment by fluorescence-activated cell sorting. Proc. Natl. Acad. Sci. USA 1979, 76, 1453–1455. [Google Scholar] [CrossRef]
- Lapaire, O.; Holzgreve, W.; Oosterwijk, J.; Brinkhaus, R.; Bianchi, D. George Schmorl: On trophoblasts in the maternal circulation. Placenta 2007, 28, 1–5. [Google Scholar] [CrossRef]
- Burton, G. Deportation of syncytial sprouts from the term human placenta. Placenta 2011, 32, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B.; Kadyrov, M.; Kingdom, J. Apoptosis and its role in the trophoblast. Am. J. Obs. Gynecol. 2006, 195, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, T. Turnover of human villous trophoblast in normal pregnancy: What do we know and what do we need to know? Placenta 2014, 35, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Weiner, C.; Weiss, M.; Zhou, H.; Syngelaki, A.; Nicolaides, K.; Dong, Y. Detection of Embryonic Trisomy 21 in the First Trimester Using Maternal Plasma Cell-Free RNA. Diagnostics 2022, 12, 1410. [Google Scholar] [CrossRef]
- Oudejans, C.; Tjoa, M.; Westerman, B.; Mulders, M.; Van Wijk, I.; Van Vugt, J. Circulating trophoblast in maternal blood. Prenat. Diagn. 2003, 23, 111–116. [Google Scholar] [CrossRef]
- Jin, J.; Menon, R. Placental exosomes: A proxy to understand pregnancy complications. Am. J. Reprod. Immunol. 2018, 79, e12788. [Google Scholar] [CrossRef]
- Taglauer, E.; Wilkins-Haug, L.; Bianchi, D. Cell-free fetal DNA in the maternal circulation as an indication of placental health and disease. Placenta 2014, 35, S64–S68. [Google Scholar] [CrossRef]
- Wataganara, T.; Chen, A.; LeShane, E.; Sullivan, L.; Borgatta, L.; Bianchi, D.; Johnson, K. Changes of cell-free fetal DNA in maternal plasma after elective termination of pregnancy. Clin. Chem. 2005, 51, 217–219. [Google Scholar] [CrossRef]
- Wright, A.; Zhou, Y.; Weier, J.; Caceres, E.; Kapidzic, M.; Tabata, T.; Kahn, M.; Nash, C.; Fisher, S. Trisomy 21 is associated with variable defects in cytotrophoblast differentiation along the invasive pathway. Am. J. Med. Gene 2004, 130A, 354–364. [Google Scholar] [CrossRef]
- Bianchi, D.; Williams, J.; Sullivan, L.; Hanson, F.; Klinger, K.; Shuber, A. PCR quantitation of fetal cells in maternal blood in normal and aneuploid pregnancies. Am. J. Hum. Genet. 1997, 61, 822–829. [Google Scholar] [CrossRef]
- Gil, M.; Accurti, V.; Santacruz, B.; Plana, M.; Nicolaides, K.H. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: Updated meta-analysis. Ultrasound Obs. Gynecol. 2017, 50, 302–314. [Google Scholar] [CrossRef]
- Svobodová, I.; Korabečná, M.; Calda, P.; Břešťák, M.; Pazourková, E.; Pospíšilová, S.; Krkavcová, M.; Novotná, M.; Hořínek, A. Differentially expressed miRNAs in trisomy 21 placentas. Prenat. Diagn. 2016, 36, 775–784. [Google Scholar] [CrossRef]
- Qureshi, F.; Jacques, S.; Johnson, M.; Hume, R.; Jr Kramer, R.; Yaron, Y.; Evans, M. Trisomy 21 placentas: Histopathological and immunohistochemical findings using proliferating cell nuclear antigen. Fetal Diagn. 1997, 12, 210–215. [Google Scholar] [CrossRef]
- Qureshi, F.; Jacques, S. Placental mesenchymal dysplasia in trisomy 21. Eur. J. Obs. Gynecol. Reprod. Biol. 2020, 247, 259. [Google Scholar] [CrossRef]
- Jauniaux, E.; Hustin, J. Chromosomally abnormal early ongoing pregnancies: Correlation of ultrasound and placental histological findings. Hum. Pathol. 1998, 29, 1195–1199. [Google Scholar] [CrossRef]
- Roberts, L.; Sebire, N.; Fowler, D.; Nicolaides, K. Histomorphological features of chorionic villi at 10–14 weeks of gestation in trisomic and chromosomally normal pregnancies. Placenta 2000, 21, 678–683. [Google Scholar] [CrossRef]
- Chen, C. Placental abnormalities and preeclampsia in trisomy 13 pregnancies. Taiwan. J. Obs. Gynecol. 2009, 48, 3–8. [Google Scholar] [CrossRef]
- Frendo, J.; Vidaud, M.; Guibourdenche, J.; Luton, D.; Muller, F.; Bellet, D.; Giovagrandi, Y.; Tarrade, A.; Porquet, D.; Blot, D.; et al. Defect of villous cytotrophoblast differentiation into syncytiotrophoblast in Down syndrom. J. Clin. Endocrinol. Metab. 2000, 85, 3700–3707. [Google Scholar]
- Massin, N.; Frendo, J.L.; Guibourdenche, J.; Luton, D.; Giovagrandi, Y.; Muller, F.; Vidaud, M.; Evain-Brion, D. Defect of syncytiotrophoblast formation and human choriogonadotropin expression in Down’s syndrome. Placenta 2001, 22, S93–S97. [Google Scholar] [CrossRef]
- Pidoux, G.; Guibourdenche, J.; Frendo, J.; Gerbaud, P.; Conti, M.; Luton, D.; Muller, F.; Evain-Brion, D. Impact of trisomy 21 on human trophoblast behaviour and function. Placenta 2004, 25, S79–S84. [Google Scholar] [CrossRef]
- Frendo, J.; Therond, P.; Bird, T.; Massin, N.; Muller, F.; Guibourdenche, J.; Luton, D.; Vidaud, M.; Anderson, W.; Evain-Brion, D. Overexpression of copper zinc superoxide dismutase impairs trophoblast cell fusion and differentiation. Endocrinology 2001, 142, 3638–3648. [Google Scholar] [CrossRef] [PubMed]
- Pidoux, G.; Gerbaud, P.; Cocquebert, M.; Segon, N.; Badet, J.; Fournier, T.; Guibourdenche, J.; Evain-Brion, D. Human trophoblast fusion and differentiation: Lessons from trisomy 21 placenta. Placenta 2012, 33, S81–S86. [Google Scholar] [CrossRef] [PubMed]
- Okmen, F.; Ekici, H.; Hortu, I.; Imamoglu, M.; Arican, D.; Akın, H.; Sagol, S. Comparison of indications and results of prenatal invasive diagnostic tests before and after the implementation of the use of cell-free fetal DNA: A tertiary referral center experience. J. Assist. Reprod. Genet. 2020, 37, 2019–2024. [Google Scholar] [CrossRef] [PubMed]
- Gullo, G.; Scaglione, M.; Buzzaccarini, G.; Laganà, A.S.; Basile, G.; Chiantera, V.; Cucinella, G.; Zaami, S. Cell-free fetal DNA and non-invasive prenatal diagnosis of chomosomopathies and pediatric monogenic diseases: A critical appraisal and medicolegal remarks. J. Pers. Med. 2023, 13, e146–e148. [Google Scholar]
- Rose, N.; Barrie, E.; Malinowski, J.; Jenkins, G.; McClain, M.; LaGrave, D.; Leung, M.; ACMG Professional Practice and Guidelines Committee. Systematic evidence-based review: The application of noninvasive prenatal screening using cell-free DNA in general-risk pregnancies. Genet. Med. 2022, 24, 1379–1391. [Google Scholar] [CrossRef]
- Barjaktarovic, M.; Korevaar, T.; Jaddoe, V.; de Rijke, Y.; Peeters, R.; Steegers, E. Human chorionic gonadotropin and risk of pre-eclampsia: Prospective population-based cohort study. Ultrasound Obstet. Gynecol. 2019, 54, 477–483. [Google Scholar] [CrossRef]
- Guibourdenche, J.; Leguy, M.C.; Tsatsaris, V. Biology and markers of preeclampsia. Ann. Biol. Clin. 2013, 71, 79–87. [Google Scholar] [CrossRef]
- Noël, L.; Guy, G.; Jones, S.; Forenc, K.; Buck, E.; Papageorghiou, A.; Thilaganathan, B. Routine first-trimester combined screening for pre-eclampsia: Pregnancy-associated plasma protein-A or placental growth factor? Ultrasound Obs. Gynecol. 2021, 58, 540–545. [Google Scholar] [CrossRef]
- Hervé, B.; Coussement, A.; Gilbert, T.; Dumont, F.; Jacques, S.; Cuisset, L.; Chicard, M.; Hizem, S.; Bourdoncle, P.; Letourneur, F.; et al. Aneuploidy: The impact of chromosome imbalance on nuclear organization and overall genome expression. Clin. Genet. 2016, 90, 35–48. [Google Scholar] [CrossRef]
- Krivega, M.; Storchova, Z. Consequences of trisomy syndromes—21 and beyond. Trends Genet. 2023, 39, 172–174. [Google Scholar] [CrossRef]
- Lim, J.; Han, Y.; Kim, H.; Kim, M.; Park, S.; Cho, Y.; Ryu, H. Integrative analyses of genes and microRNA expressions in human trisomy 21 placentas. BMC Med. Genom. 2018, 11, 46. [Google Scholar] [CrossRef]
- Ghafourian, M.; Mahdavi, R.; Jonoush, Z.; Sadeghj, M.; Ghadiri, N.; Farzaneh, M.; Salehi, A. The implications of exosomes in pregnancy: Emerging as new diagnostic markers and therapeutics targets. Cell. Commun. Signal. 2022, 20, 51. [Google Scholar] [CrossRef]
- Adams, A.; Guedj, F.; Bianchi, D. Placental development and function in trisomy 21 and mouse models of Down syndrome: Clues for studying mechanisms underlying atypical development. Placenta 2020, 89, 58–66. [Google Scholar] [CrossRef]
- Ganguly, B.; Kadam, N. Triplication of HSA21 on alterations in structure and function of mitochondria. Mitochondrion 2022, 65, 88–101. [Google Scholar] [CrossRef]
- Tan, K.; Lee, H.; Cheah, P.; Ling, K. Mitochondrial Dysfunction in Down Syndrome: From Pathology to Therapy. Neuroscience 2023, 511, 1–12. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guibourdenche, J.; Leguy, M.-C.; Pidoux, G.; Hebert-Schuster, M.; Laguillier, C.; Anselem, O.; Grangé, G.; Bonnet, F.; Tsatsaris, V. Biochemical Screening for Fetal Trisomy 21: Pathophysiology of Maternal Serum Markers and Involvement of the Placenta. Int. J. Mol. Sci. 2023, 24, 7669. https://doi.org/10.3390/ijms24087669
Guibourdenche J, Leguy M-C, Pidoux G, Hebert-Schuster M, Laguillier C, Anselem O, Grangé G, Bonnet F, Tsatsaris V. Biochemical Screening for Fetal Trisomy 21: Pathophysiology of Maternal Serum Markers and Involvement of the Placenta. International Journal of Molecular Sciences. 2023; 24(8):7669. https://doi.org/10.3390/ijms24087669
Chicago/Turabian StyleGuibourdenche, Jean, Marie-Clémence Leguy, Guillaume Pidoux, Marylise Hebert-Schuster, Christelle Laguillier, Olivia Anselem, Gilles Grangé, Fidéline Bonnet, and Vassilis Tsatsaris. 2023. "Biochemical Screening for Fetal Trisomy 21: Pathophysiology of Maternal Serum Markers and Involvement of the Placenta" International Journal of Molecular Sciences 24, no. 8: 7669. https://doi.org/10.3390/ijms24087669