
Drug Discovery Targeting Post-Translational Modifications in Response to DNA Damages Induced by Space Radiation


Abstract
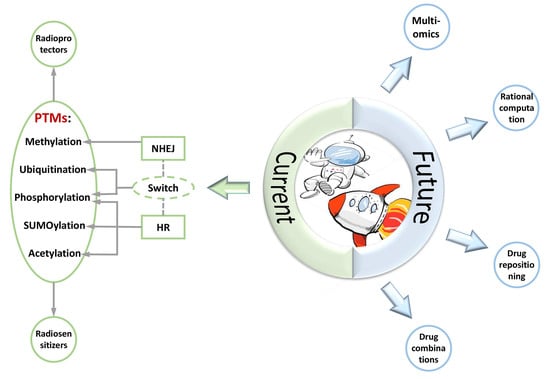
:
1. Introduction
2. Literature Search and Curation
3. PTMs in the Choice of DNA Repair Pathways
4. Targeting Essential Phosphorylation Factors for Regulating DDR
5. Ubiquitylation and Its Roles in Regulating DNA Repair
6. DNA Damage Repair Modulated by Other PTMs
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Alt-EJ | alternative end joining |
| DDR | DNA damage response |
| DSB | double-strand break |
| HR | homologous recombination |
| ICL | interstrand cross-link |
| IR | ionizing radiation |
| Kcr | lysine crotonylation |
| LET | linear energy transfer |
| NHEJ | non-homologous end joining |
| NO | nitric oxide |
| OGT | O-GlcNAc transferase |
| PARylation | poly ADP-ribosylation |
| PROTAC | proteolysis-targeting chimera |
| PTM | post-translational modification |
| R&D | research and development |
| RT | radiation therapy |
| SSA | single-strand annealing |
| ssDNA | single-strand DNA |
| US FDA | United States Food and Drug Administration |
References
- Ramos, R.L.; Carante, M.P.; Ferrari, A.; Sala, P.; Vercesi, V.; Ballarini, F. A Mission to Mars: Prediction of GCR Doses and Comparison with Astronaut Dose Limits. Int. J. Mol. Sci. 2023, 24, 2328. [Google Scholar] [CrossRef] [PubMed]
- Drago-Ferrante, R.; Di Fiore, R.; Karouia, F.; Subbannayya, Y.; Das, S.; Aydogan Mathyk, B.; Arif, S.; Guevara-Cerdán, A.P.; Seylani, A.; Galsinh, A.S.; et al. Extraterrestrial Gynecology: Could Spaceflight Increase the Risk of Developing Cancer in Female Astronauts? An Updated Review. Int. J. Mol. Sci. 2022, 23, 7465. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.K.; Liu, B.; Hinshaw, R.G.; Park, M.-A.; Wang, S.; Dubey, S.; Liu, G.G.; Shi, Q.; Holton, P.; Reiser, V.; et al. Long-Term Sex- and Genotype-Specific Effects of 56Fe Irradiation on Wild-Type and APPswe/PS1dE9 Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 13305. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N.; Sato, T. Cataractogenesis following high-LET radiation exposure. Mutat. Res. 2016, 770, 262–291. [Google Scholar] [CrossRef] [PubMed]
- Rudobeck, E.; Bellone, J.A.; Szücs, A.; Bonnick, K.; Mehrotra-Carter, S.; Badaut, J.; Nelson, G.A.; Hartman, R.E.; Vlkolinský, R. Low-dose proton radiation effects in a transgenic mouse model of Alzheimer’s disease—Implications for space travel. PLoS ONE 2017, 12, e0186168. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.A.; Sasi, S.P.; Onufrak, J.; Natarajan, M.; Manickam, K.; Schwab, J.; Muralidharan, S.; Peterson, L.E.; Alekseyev, Y.O.; Yan, X.; et al. Low-dose radiation affects cardiac physiology: Gene networks and molecular signaling in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1947–H1963. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Blakely, E.A.; Burma, S.; Fornace, A.J.; Gerson, S.; Hlatky, L.; Kirsch, D.G.; Luderer, U.; Shay, J.; Wang, Y.; et al. Concepts and challenges in cancer risk prediction for the space radiation environment. Life Sci. Space Res. 2015, 6, 92–103. [Google Scholar] [CrossRef]
- Cucinotta, F.A. Flying without a Net: Space Radiation Cancer Risk Predictions without a Gamma-ray Basis. Int. J. Mol. Sci. 2022, 23, 4324. [Google Scholar] [CrossRef]
- Poignant, F.; Plante, I.; Patel, Z.S.; Huff, J.L.; Slaba, T.C. Geometrical Properties of the Nucleus and Chromosome Intermingling Are Possible Major Parameters of Chromosome Aberration Formation. Int. J. Mol. Sci. 2022, 23, 8638. [Google Scholar] [CrossRef]
- Yachi, Y.; Matsuya, Y.; Yoshii, Y.; Fukunaga, H.; Date, H.; Kai, T. An Analytical Method for Quantifying the Yields of DNA Double-Strand Breaks Coupled with Strand Breaks by γ-H2AX Focus Formation Assay Based on Track-Structure Simulation. Int. J. Mol. Sci. 2023, 24, 1386. [Google Scholar] [CrossRef]
- Hu, A.; Zhou, W.; Wu, Z.; Zhang, H.; Li, J.; Qiu, R. Modeling of DNA Damage Repair and Cell Response in Relation to p53 System Exposed to Ionizing Radiation. Int. J. Mol. Sci. 2022, 23, 11323. [Google Scholar] [CrossRef] [PubMed]
- Hirose, E.; Noguchi, M.; Ihara, T.; Yokoya, A. Mitochondrial Metabolism in X-Irradiated Cells Undergoing Irreversible Cell-Cycle Arrest. Int. J. Mol. Sci. 2023, 24, 1833. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Bing, Z.; Zhang, Y.; Ao, B.; Zhang, S.; Ye, C.; He, J.; Ding, N.; Ye, W.; Xiong, J.; et al. Quantitative proteomic analysis for radiation-induced cell cycle suspension in 92-1 melanoma cell line. J. Radiat. Res. 2013, 54, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Albi, E.; Cataldi, S.; Lazzarini, A.; Codini, M.; Beccari, T.; Ambesi-Impiombato, F.S.; Curcio, F. Radiation and Thyroid Cancer. Int. J. Mol. Sci. 2017, 18, 911. [Google Scholar] [CrossRef]
- Nie, Q.; Huan, X.; Kang, J.; Yin, J.; Zhao, J.; Li, Y.; Zhang, Z. MG149 Inhibits MOF-Mediated p53 Acetylation to Attenuate X-ray Radiation-Induced Apoptosis in H9c2 Cells. Radiat. Res. 2022, 198, 590–598. [Google Scholar] [CrossRef]
- Tinganelli, W.; Luoni, F.; Durante, M. What can space radiation protection learn from radiation oncology? Life Sci. Space Res. 2021, 30, 82–95. [Google Scholar] [CrossRef]
- Putt, K.S.; Du, Y.; Fu, H.; Zhang, Z.-Y. High-throughput screening strategies for space-based radiation countermeasure discovery. Life Sci. Space Res. 2022, 35, 88–104. [Google Scholar] [CrossRef]
- Cheema, A.K.; Mehta, K.Y.; Fatanmi, O.O.; Wise, S.Y.; Hinzman, C.P.; Wolff, J.; Singh, V.K. A Metabolomic and Lipidomic Serum Signature from Nonhuman Primates Administered with a Promising Radiation Countermeasure, Gamma-Tocotrienol. Int. J. Mol. Sci. 2017, 19, 79. [Google Scholar] [CrossRef]
- Gan, L.; Wang, Z.; Si, J.; Zhou, R.; Sun, C.; Liu, Y.; Ye, Y.; Zhang, Y.; Liu, Z.; Zhang, H. Protective effect of mitochondrial-targeted antioxidant MitoQ against iron ion 56Fe radiation induced brain injury in mice. Toxicol. Appl. Pharmacol. 2018, 341, 1–7. [Google Scholar] [CrossRef]
- Yang, P.; Luo, X.; Li, J.; Zhang, T.; Gao, X.; Hua, J.; Li, Y.; Ding, N.; He, J.; Zhang, Y.; et al. Ionizing Radiation Upregulates Glutamine Metabolism and Induces Cell Death via Accumulation of Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2021, 2021, 5826932. [Google Scholar] [CrossRef]
- Moreno-Villanueva, M.; Wong, M.; Lu, T.; Zhang, Y.; Wu, H. Interplay of space radiation and microgravity in DNA damage and DNA damage response. NPJ Microgravity 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ge, C.; Yang, L.; Wang, R.; Lu, Y.; Gao, Y.; Li, Z.; Wu, Y.; Zheng, X.; Wang, Z.; et al. CBLB502, an Agonist of Toll-Like Receptor 5, has Antioxidant and Scavenging Free Radicals Activities in vitro. Int. J. Biol. Macromol. 2016, 82, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Hosseinimehr, S.J. The protective effects of trace elements against side effects induced by ionizing radiation. Radiat. Oncol. J. 2015, 33, 66–74. [Google Scholar] [CrossRef]
- Burns, F.J.; Tang, M.; Frenkel, K.; Nádas, A.; Wu, F.; Uddin, A.; Zhang, R. Induction and prevention of carcinogenesis in rat skin exposed to space radiation. Radiat. Environ. Biophys. 2007, 46, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Tan, Y.; Huang, C.Q.; Xu, M.C.; Li, Q.; Pan, D.; Zhao, B.Q.; Hu, B.R. MMP Inhibitor Ilomastat Improves Survival of Mice Exposed to γ-Irradiation. Biomed. Environ. Sci. BES 2018, 31, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Clemente, A.; Sonnante, G.; Domoney, C. Bowman-Birk inhibitors from legumes and human gastrointestinal health: Current status and perspectives. Curr. Protein Pept. Sci. 2011, 12, 358–373. [Google Scholar] [CrossRef]
- Kennedy, A.R.; Zhou, Z.; Donahue, J.J.; Ware, J.H. Protection against adverse biological effects induced by space radiation by the Bowman-Birk inhibitor and antioxidants. Radiat. Res. 2006, 166, 327–332. [Google Scholar] [CrossRef]
- Garg, S.; Garg, T.K.; Wise, S.Y.; Fatanmi, O.O.; Miousse, I.R.; Savenka, A.V.; Basnakian, A.G.; Singh, V.K.; Hauer-Jensen, M. Effects of Gamma-Tocotrienol on Intestinal Injury in a GI-Specific Acute Radiation Syndrome Model in Nonhuman Primate. Int. J. Mol. Sci. 2022, 23, 4643. [Google Scholar] [CrossRef]
- Singh, V.K.; Hauer-Jensen, M. γ-Tocotrienol as a Promising Countermeasure for Acute Radiation Syndrome: Current Status. Int. J. Mol. Sci. 2016, 17, 663. [Google Scholar] [CrossRef]
- Lulli, M.; Witort, E.; Papucci, L.; Torre, E.; Schiavone, N.; Dal Monte, M.; Capaccioli, S. Coenzyme Q10 protects retinal cells from apoptosis induced by radiation in vitro and in vivo. J. Radiat. Res. 2012, 53, 695–703. [Google Scholar] [CrossRef]
- Li, J.; Xu, J.; Xu, W.; Qi, Y.; Lu, Y.; Qiu, L.; Hu, Z.; Chu, Z.; Chai, Y.; Zhang, J. Protective Effects of Hong Shan Capsule against Lethal Total-Body Irradiation-Induced Damage in Wistar Rats. Int. J. Mol. Sci. 2015, 16, 18938–18955. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, L.; Xing, Y.; Wang, Y.; Du, L.; Xu, C.; Cao, J.; Wang, Q.; Fan, S.; Liu, Q.; et al. Radioprotective and antioxidant effect of resveratrol in hippocampus by activating Sirt1. Int. J. Mol. Sci. 2014, 15, 5928–5939. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, M.; Matsushita, N. DNA Damage Response Regulation by Histone Ubiquitination. Int. J. Mol. Sci. 2022, 23, 8187. [Google Scholar] [CrossRef] [PubMed]
- Casari, E.; Rinaldi, C.; Marsella, A.; Gnugnoli, M.; Colombo, C.V.; Bonetti, D.; Longhese, M.P. Processing of DNA Double-Strand Breaks by the MRX Complex in a Chromatin Context. Front. Mol. Biosci. 2019, 6, 43. [Google Scholar] [CrossRef]
- Uckelmann, M.; Sixma, T.K. Histone ubiquitination in the DNA damage response. DNA Repair 2017, 56, 92–101. [Google Scholar] [CrossRef]
- Mladenov, E.; Paul-Konietzko, K.; Mladenova, V.; Stuschke, M.; Iliakis, G. Increased Gene Targeting in Hyper-Recombinogenic LymphoBlastoid Cell Lines Leaves Unchanged DSB Processing by Homologous Recombination. Int. J. Mol. Sci. 2022, 23, 9180. [Google Scholar] [CrossRef]
- Argunhan, B.; Iwasaki, H.; Tsubouchi, H. Post-translational modification of factors involved in homologous recombination. DNA Repair 2021, 104, 103114. [Google Scholar] [CrossRef]
- Yu, F.; Wei, J.; Cui, X.; Yu, C.; Ni, W.; Bungert, J.; Wu, L.; He, C.; Qian, Z. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic. Acids Res. 2021, 49, 5779–5797. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Tang, M.; Li, S.; Chen, J. Ubiquitylation in DNA double-strand break repair. DNA Repair 2021, 103, 103129. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Yuan, W.; Jacob, Y. The Role of the TSK/TONSL-H3.1 Pathway in Maintaining Genome Stability in Multicellular Eukaryotes. Int. J. Mol. Sci. 2022, 23, 9029. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly (ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, D. Repair pathway choice for double-strand breaks. Essays Biochem. 2020, 64, 765–777. [Google Scholar] [CrossRef]
- Zierhut, C.; Diffley, J.F.X. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008, 27, 1875–1885. [Google Scholar] [CrossRef]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef]
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.E.; Li, Y.; Wu-Baer, F.; Chait, B.T.; Baer, R.; Yan, H.; Gottesman, M.E.; Gautier, J. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell 2013, 49, 657–667. [Google Scholar] [CrossRef]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Chapman, J.R.; Barral, P.; Vannier, J.-B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 2013, 49, 858–871. [Google Scholar] [CrossRef]
- Spagnolo, L.; Rivera-Calzada, A.; Pearl, L.H.; Llorca, O. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell 2006, 22, 511–519. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Deng, L.; Nguyen, S.C.; Zhao, X.; Maulion, C.D.; Shao, C.; Tischfield, J.A. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 3297–3310. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Díaz, C.; Orthwein, A.; Leung, C.C.Y.; Huang, H.; Landry, M.-C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef]
- Barazas, M.; Annunziato, S.; Pettitt, S.J.; de Krijger, I.; Ghezraoui, H.; Roobol, S.J.; Lutz, C.; Frankum, J.; Song, F.F.; Brough, R.; et al. The CST Complex Mediates End Protection at Double-Strand Breaks and Promotes PARP Inhibitor Sensitivity in BRCA1-Deficient Cells. Cell Rep. 2018, 23, 2107–2118. [Google Scholar] [CrossRef]
- Turan, V.; Oktay, K. BRCA-related ATM-mediated DNA double-strand break repair and ovarian aging. Hum. Reprod. Update 2020, 26, 43–57. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Rahib, L.; Lyons, E.; De Arbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sohal, D.P.S.; et al. Outcomes in Patients With Pancreatic Adenocarcinoma With Genetic Mutations in DNA Damage Response Pathways: Results From the Know Your Tumor Program. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, S. Targeting DNA Double-Strand Break (DSB) Repair to Counteract Tumor Radio-resistance. Curr. Drug Targets 2019, 20, 891–902. [Google Scholar] [CrossRef]
- Zhu, Z.; Chung, W.-H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.-H.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Wan, L.; Busygina, V.; Kwon, Y.; Allen, J.A.; Li, X.; Kunz, R.C.; Kubota, K.; Wang, B.; Sung, P.; et al. Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol. Cell 2009, 36, 393–404. [Google Scholar] [CrossRef]
- Tsubouchi, H.; Roeder, G.S. Budding yeast Hed1 down-regulates the mitotic recombination machinery when meiotic recombination is impaired. Genes. Dev. 2006, 20, 1766–1775. [Google Scholar] [CrossRef]
- Dhingra, N.; Zhao, X. Advances in SUMO-based regulation of homologous recombination. Curr. Opin. Genet. Dev. 2021, 71, 114–119. [Google Scholar] [CrossRef]
- Hariharasudhan, G.; Jeong, S.-Y.; Kim, M.-J.; Jung, S.M.; Seo, G.; Moon, J.-R.; Lee, S.; Chang, I.-Y.; Kee, Y.; You, H.J.; et al. TOPORS-mediated RAD51 SUMOylation facilitates homologous recombination repair. Nucleic Acids Res. 2022, 50, 1501–1516. [Google Scholar] [CrossRef]
- Ferretti, L.P.; Himmels, S.-F.; Trenner, A.; Walker, C.; von Aesch, C.; Eggenschwiler, A.; Murina, O.; Enchev, R.I.; Peter, M.; Freire, R.; et al. Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat. Commun. 2016, 7, 12628. [Google Scholar] [CrossRef]
- Ismail, I.H.; Gagné, J.-P.; Genois, M.-M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.-Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 2015, 17, 1446–1457. [Google Scholar] [CrossRef]
- Lafranchi, L.; de Boer, H.R.; de Vries, E.G.E.; Ong, S.-E.; Sartori, A.A.; van Vugt, M.A.T.M. APC/C(Cdh1) controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J. 2014, 33, 2860–2879. [Google Scholar] [CrossRef]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Cornwell, M.; Beli, P.; Jackson, S.P. Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting DNA repair. Nat. Cell Biol. 2015, 17, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.R.; Clifford, G.; Bonnet, C.; Groth, A.; Wilson, M.D.; Chapman, J.R. BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination. Nature 2021, 596, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Walser, F.; Mulder, M.P.C.; Bragantini, B.; Burger, S.; Gubser, T.; Gatti, M.; Botuyan, M.V.; Villa, A.; Altmeyer, M.; Neri, D.; et al. Ubiquitin Phosphorylation at Thr12 Modulates the DNA Damage Response. Mol. Cell 2020, 80, 423–436.e9. [Google Scholar] [CrossRef] [PubMed]
- Callen, E.; Di Virgilio, M.; Kruhlak, M.J.; Nieto-Soler, M.; Wong, N.; Chen, H.-T.; Faryabi, R.B.; Polato, F.; Santos, M.; Starnes, L.M.; et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013, 153, 1266–1280. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Fong, K.-W.; Wang, J.; Wang, W.; Chen, J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013, 288, 11135–11143. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.-L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef]
- Chen, B.P.C.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Löbrich, M.; Shiloh, Y.; Chen, D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle Georget. Tex. 2010, 9, 472–478. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef]
- Kery, M.; Papandreou, I. Emerging strategies to target cancer metabolism and improve radiation therapy outcomes. Br. J. Radiol. 2020, 93, 20200067. [Google Scholar] [CrossRef] [PubMed]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Morita, A.; Tatsuta, S.; Kanamaru, M.; Sakaue, M.; Ueda, K.; Shono, M.; Fujita, R.; Wang, B.; Hosoi, Y.; et al. Isorhamnetin Promotes 53BP1 Recruitment through the Enhancement of ATM Phosphorylation and Protects Mice from Radiation Gastrointestinal Syndrome. Genes 2021, 12, 1514. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fischer, T.; Katzer, A.; Sisario, D.; Korsa, T.; Steussloff, G.; Sukhorukov, V.L.; Flentje, M. Opposite effects of the triple target (DNA-PK/PI3K/mTOR) inhibitor PI-103 on the radiation sensitivity of glioblastoma cell lines proficient and deficient in DNA-PKcs. BMC Cancer 2021, 21, 1201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, H.; Hao, J.; Wu, Y.; Yang, B. Inhibition of DNA-PKcs activity re-sensitizes uveal melanoma cells to radio- and chemotherapy. Biochem. Biophys. Res. Commun. 2020, 522, 639–646. [Google Scholar] [CrossRef]
- Ciszewski, W.M.; Tavecchio, M.; Dastych, J.; Curtin, N.J. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res. Treat. 2014, 143, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Zhang, Y.; Lai, J.; Du, Z.; Gao, J.; Yang, S.; Gorityala, S.; Xiong, X.; Deng, O.; Ma, Z.; Yan, C.; et al. Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget 2016, 7, 34688–34702. [Google Scholar] [CrossRef] [PubMed]
- Udayakumar, D.; Pandita, R.K.; Horikoshi, N.; Liu, Y.; Liu, Q.; Wong, K.-K.; Hunt, C.R.; Gray, N.S.; Minna, J.D.; Pandita, T.K.; et al. Torin2 Suppresses Ionizing Radiation-Induced DNA Damage Repair. Radiat. Res. 2016, 185, 527–538. [Google Scholar] [CrossRef]
- Chen, J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000, 60, 5037–5039. [Google Scholar]
- Hu, S.; Hui, Z.; Duan, J.; Garrido, C.; Xie, T.; Ye, X.-Y. Discovery of small-molecule ATR inhibitors for potential cancer treatment: A patent review from 2014 to present. Expert Opin. Ther. Pat. 2022, 32, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Cho, W.K.; Park, S.; Shin, S.-W.; Park, W.; Kim, H.; Choi, D.H. Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation. Int. J. Mol. Sci. 2020, 21, 2691. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.S.; Huang, S.-B.; Bedolla, R.G.; Rivas, P.; Basler, J.W.; Swanson, G.P.; Hui-Ming Huang, T.; Narayanasamy, G.; Papanikolaou, N.; Miyamoto, H.; et al. Suppression of ribosomal protein RPS6KB1 by Nexrutine increases sensitivity of prostate tumors to radiation. Cancer Lett. 2018, 433, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Fan, G.; Deng, C.; Wu, L. Mir-4429 sensitized cervical cancer cells to irradiation by targeting RAD51. J. Cell Physiol. 2020, 235, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, H.; Chen, X.; Xiong, J.; Song, Z. Silencing UCHL3 enhances radio-sensitivity of non-small cell lung cancer cells by inhibiting DNA repair. Aging 2021, 13, 14277–14288. [Google Scholar] [CrossRef]
- Du, L.-Q.; Du, X.-Q.; Bai, J.-Q.; Wang, Y.; Yang, Q.-S.; Wang, X.-C.; Zhao, P.; Wang, H.; Liu, Q.; Fan, F.-Y. Methotrexate-mediated inhibition of RAD51 expression and homologous recombination in cancer cells. J. Cancer Res. Clin. Oncol. 2012, 138, 811–818. [Google Scholar] [CrossRef]
- Gemenetzidis, E.; Gammon, L.; Biddle, A.; Emich, H.; Mackenzie, I.C. Invasive oral cancer stem cells display resistance to ionising radiation. Oncotarget 2015, 6, 43964–43977. [Google Scholar] [CrossRef]
- Tsukuda, T.; Fleming, A.B.; Nickoloff, J.A.; Osley, M.A. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 2005, 438, 379–383. [Google Scholar] [CrossRef]
- Zgheib, O.; Pataky, K.; Brugger, J.; Halazonetis, T.D. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell Biol. 2009, 29, 1050–1058. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.A.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.-N.; Moi, S.-H.; Hou, M.-F.; Luo, C.-W.; Chen, F.-M.; Pan, M.-R. RNF8-CDH1 Co-Expression Predicts Clinical Benefit of Chemoradiotherapy in Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.A.; Berti, M.; Walser, F.; Raso, M.C.; Schmid, F.; Krietsch, J.; Stoy, H.; Zwicky, K.; Ursich, S.; Freire, R.; et al. Histone Ubiquitination by the DNA Damage Response Is Required for Efficient DNA Replication in Unperturbed S Phase. Mol. Cell 2018, 71, 897–910.e8. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE Syndrome Protein Mediates a Ubiquitin-Dependent Signaling Cascade at Sites of DNA Damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef]
- Wang, F.-C.; Peng, B.; Ren, T.-T.; Liu, S.-P.; Du, J.-R.; Chen, Z.-H.; Zhang, T.-T.; Gu, X.; Li, M.; Cao, S.-L.; et al. A 1,2,3-Triazole Derivative of Quinazoline Exhibits Antitumor Activity by Tethering RNF168 to SQSTM1/P62. J. Med. Chem. 2022, 65, 15028–15047. [Google Scholar] [CrossRef]
- Ross, A.-L.; Simpson, L.J.; Sale, J.E. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005, 33, 1280–1289. [Google Scholar] [CrossRef]
- Saha, P.; Mandal, T.; Talukdar, A.D.; Kumar, D.; Kumar, S.; Tripathi, P.P.; Wang, Q.-E.; Srivastava, A.K. DNA polymerase eta: A potential pharmacological target for cancer therapy. J. Cell Physiol. 2021, 236, 4106–4120. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; Wang, F.; Temviriyanukul, P.; Ma, X.; Tu, Y.; Lv, L.; Lin, Y.-F.; Huang, M.; Zhang, T.; et al. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 2015, 43, 8325–8339. [Google Scholar] [CrossRef]
- Guo, C.; Tang, T.-S.; Bienko, M.; Parker, J.L.; Bielen, A.B.; Sonoda, E.; Takeda, S.; Ulrich, H.D.; Dikic, I.; Friedberg, E.C. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell Biol. 2006, 26, 8892–8900. [Google Scholar] [CrossRef]
- Alimova, I.; Wang, D.; Danis, E.; Pierce, A.; Donson, A.; Serkova, N.; Madhavan, K.; Lakshmanachetty, S.; Balakrishnan, I.; Foreman, N.K.; et al. Targeting the TP53/MDM2 axis enhances radiation sensitivity in atypical teratoid rhabdoid tumors. Int. J. Oncol. 2022, 60, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhu, Y.; Zhu, J.; Tao, J.; Wei, X.; Wo, Y.; Hou, H. Primary resistance to first-generation EGFR-TKIs induced by MDM2 amplification in NSCLC. Mol. Med. Camb. Mass 2020, 26, 66. [Google Scholar] [CrossRef]
- Kim, B.H.; Kim, Y.J.; Kim, M.-H.; Na, Y.R.; Jung, D.; Seok, S.H.; Kim, J.; Kim, H.J. Identification of FES as a Novel Radiosensitizing Target in Human Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Decaudin, D.; Frisch Dit Leitz, E.; Nemati, F.; Tarin, M.; Naguez, A.; Zerara, M.; Marande, B.; Vivet-Noguer, R.; Halilovic, E.; Fabre, C.; et al. Preclinical evaluation of drug combinations identifies co-inhibition of Bcl-2/XL/W and MDM2 as a potential therapy in uveal melanoma. Eur. J. Cancer 2020, 126, 93–103. [Google Scholar] [CrossRef]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Jian Ma, C.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Park, J.-W.; Park, J.-E.; Kim, S.-R.; Sim, M.-K.; Kang, C.-M.; Kim, K.S. Metformin alleviates ionizing radiation-induced senescence by restoring BARD1-mediated DNA repair in human aortic endothelial cells. Exp. Gerontol. 2022, 160, 111706. [Google Scholar] [CrossRef]
- Liu, G.; Lim, D.; Cai, Z.; Ding, W.; Tian, Z.; Dong, C.; Zhang, F.; Guo, G.; Wang, X.; Zhou, P.; et al. The Valproate Mediates Radio-Bidirectional Regulation Through RFWD3-Dependent Ubiquitination on Rad51. Front. Oncol. 2021, 11, 646256. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Blackburn, T.J.; Cook, S.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Hewitt, L.; Huberman, K.; McNeill, H.V.; Newell, D.R.; et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS ONE 2012, 7, e45539. [Google Scholar] [CrossRef]
- Wang, X.; Wei, L.; Cramer, J.M.; Leibowitz, B.J.; Judge, C.; Epperly, M.; Greenberger, J.; Wang, F.; Li, L.; Stelzner, M.G.; et al. Pharmacologically blocking p53-dependent apoptosis protects intestinal stem cells and mice from radiation. Sci. Rep. 2015, 5, 8566. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lin, C.; Dong, H.; Piao, Z.; Jin, C.; Han, H.; Jin, D. Targeting MALAT1 induces DNA damage and sensitize non-small cell lung cancer cells to cisplatin by repressing BRCA1. Cancer Chemother. Pharmacol. 2020, 86, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Classen, S.; Rahlf, E.; Jungwirth, J.; Albers, N.; Hebestreit, L.P.; Zielinski, A.; Poole, L.; Groth, M.; Koch, P.; Liehr, T.; et al. Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance. Int. J. Mol. Sci. 2022, 23, 13363. [Google Scholar] [CrossRef]
- Affandi, T.; Ohm, A.M.; Gaillard, D.; Haas, A.; Reyland, M.E. Tyrosine kinase inhibitors protect the salivary gland from radiation damage by increasing DNA double-strand break repair. J. Biol. Chem. 2021, 296, 100401. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Peng, R.; Yu, H.; Chen, Z.; Wang, S.; Wang, Z.; Dong, S.; Li, W.; Jiang, Q.; Li, F.; et al. Dimethyl Sulfoxide Attenuates Radiation-Induced Testicular Injury through Facilitating DNA Double-Strand Break Repair. Oxid. Med. Cell Longev. 2022, 2022, 9137812. [Google Scholar] [CrossRef]
- Chang, S.; Hu, L.; Xu, Y.; Li, X.; Ma, L.; Feng, X.; Wang, J.; Zhang, C.; Wang, S. Inorganic Nitrate Alleviates Total Body Irradiation-Induced Systemic Damage by Decreasing Reactive Oxygen Species Levels. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 945–957. [Google Scholar] [CrossRef]
- Lee, V.; Gober, M.D.; Bashir, H.; O’Day, C.; Blair, I.A.; Mesaros, C.; Weng, L.; Huang, A.; Chen, A.; Tang, R.; et al. Voriconazole enhances UV-induced DNA damage by inhibiting catalase and promoting oxidative stress. Exp. Dermatol. 2020, 29, 29–38. [Google Scholar] [CrossRef]
- Liu, C.; Vyas, A.; Kassab, M.A.; Singh, A.K.; Yu, X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017, 45, 8129–8141. [Google Scholar] [CrossRef]
- Prokhorova, E.; Zobel, F.; Smith, R.; Zentout, S.; Gibbs-Seymour, I.; Schützenhofer, K.; Peters, A.; Groslambert, J.; Zorzini, V.; Agnew, T.; et al. Serine-linked PARP1 auto-modification controls PARP inhibitor response. Nat. Commun. 2021, 12, 4055. [Google Scholar] [CrossRef]
- Brown, J.S.; Lukashchuk, N.; Sczaniecka-Clift, M.; Britton, S.; le Sage, C.; Calsou, P.; Beli, P.; Galanty, Y.; Jackson, S.P. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015, 11, 704–714. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Hu, Z.; Shen, Y.; Jiang, Y.; Pan, P.; Hou, T.; Pan, Z.-Q.; Huang, J.; Sun, Y. Gossypol inhibits cullin neddylation by targeting SAG-CUL5 and RBX1-CUL1 complexes. Neoplasia 2020, 22, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; You, X.; Xu, T.; Chen, Q.; Li, H.; Dou, L.; Sun, Y.; Xiong, X.; Meredith, M.A.; Sun, Y. PD-L1 induction via the MEK-JNK-AP1 axis by a neddylation inhibitor promotes cancer-associated immunosuppression. Cell Death Dis. 2022, 13, 844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zheng, L.; Hu, K.; Wang, X.; Zhang, R.; Zou, Y.; Zhong, L.; Wang, S.; Wu, Y.; Kang, T. SUMOylation stabilizes hSSB1 and enhances the recruitment of NBS1 to DNA damage sites. Signal Transduct. Target. Ther. 2020, 5, 80. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, Q.; Liu, H.; He, F.-J.; Liu, J.-H.; Zhou, Y.-Y.; Zeng, M.-T.; Pei, Q.; Zhu, H. SUMOylation of methyltransferase-like 3 facilitates colorectal cancer progression by promoting circ_0000677 in an m6 A-dependent manner. J. Gastroenterol. Hepatol. 2022, 37, 700–713. [Google Scholar] [CrossRef]
- Traver, G.; Sekhar, K.R.; Crooks, P.A.; Keeney, D.S.; Freeman, M.L. Targeting NPM1 in irradiated cells inhibits NPM1 binding to RAD51, RAD51 foci formation and radiosensitizes NSCLC. Cancer Lett. 2021, 500, 220–227. [Google Scholar] [CrossRef]
- Ping, X.; Stark, J.M. O-GlcNAc transferase is important for homology-directed repair. DNA Repair 2022, 119, 103394. [Google Scholar] [CrossRef]
- He, N.; Ma, D.; Tan, Y.; Liu, M. Upregulation of O-GlcNAc transferase is involved in the pathogenesis of acute myeloid leukemia. Asia Pac. J. Clin. Oncol. 2022, 18, e318–e328. [Google Scholar] [CrossRef]
- Semlow, D.R.; Zhang, J.; Budzowska, M.; Drohat, A.; Walter, J.C. Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell 2016, 167, 498–511.e14. [Google Scholar] [CrossRef]
- Li, N.; Wang, J.; Wallace, S.S.; Chen, J.; Zhou, J.; D’Andrea, A.D. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 2020, 48, 3014–3028. [Google Scholar] [CrossRef]
- Wang, Q.; Li, Z.; Yang, J.; Peng, S.; Zhou, Q.; Yao, K.; Cai, W.; Xie, Z.; Qin, F.; Li, H.; et al. Loss of NEIL3 activates radiotherapy resistance in the progression of prostate cancer. Cancer Biol. Med. 2021, 19, 1193–1210. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, L.; Shi, S.; Wu, S.; Meng, R.; Chen, H.; Jiang, Z. Deficiency of NEIL3 Enhances the Chemotherapy Resistance of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 4098. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Haskins, J.S.; Su, C.; Allum, A.; Haskins, A.H.; Salinas, V.A.; Sunada, S.; Inoue, T.; Aizawa, Y.; Uesaka, M.; et al. In vitro screening of radioprotective properties in the novel glucosylated flavonoids. Int. J. Mol. Med. 2016, 38, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Machour, F.E.; Ayoub, N. Transcriptional Regulation at DSBs: Mechanisms and Consequences. Trends Genet. TIG 2020, 36, 981–997. [Google Scholar] [CrossRef]
- Liu, X.; Wei, W.; Liu, Y.; Yang, X.; Wu, J.; Zhang, Y.; Zhang, Q.; Shi, T.; Du, J.X.; Zhao, Y.; et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Sabari, B.R.; Tang, Z.; Huang, H.; Yong-Gonzalez, V.; Molina, H.; Kong, H.E.; Dai, L.; Shimada, M.; Cross, J.R.; Zhao, Y.; et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 2015, 58, 203–215. [Google Scholar] [CrossRef]
- Mooring, S.R.; Jin, H.; Devi, N.S.; Jabbar, A.A.; Kaluz, S.; Liu, Y.; Van Meir, E.G.; Wang, B. Design and synthesis of novel small-molecule inhibitors of the hypoxia inducible factor pathway. J. Med. Chem. 2011, 54, 8471–8489. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-X.; Wei, B.-F.; Zhang, X.; Gong, Y.-P.; Ma, L.-Y.; Zhao, W. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 2021, 209, 112861. [Google Scholar] [CrossRef]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Guo, Z.; Liu, F.; Zhang, W.; Ke, X.; Su, Y.; Wang, M.; Yao, Y.; et al. ATM Expression Is Elevated in Established Radiation-Resistant Breast Cancer Cells and Improves DNA Repair Efficiency. Int. J. Biol. Sci. 2020, 16, 1096–1106. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Scholz, A.; Haendler, B. Targeting DNA Damage Response in Prostate and Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8273. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lee, J.-H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef]
- Dylgjeri, E.; Kothari, V.; Shafi, A.A.; Semenova, G.; Gallagher, P.T.; Guan, Y.F.; Pang, A.; Goodwin, J.F.; Irani, S.; McCann, J.J.; et al. A Novel Role for DNA-PK in Metabolism by Regulating Glycolysis in Castration-Resistant Prostate Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Efimova, E.V.; Ramamurthy, A.; Kron, S.J. Repair-independent functions of DNA-PKcs protect irradiated cells from mitotic slippage and accelerated senescence. J. Cell Sci. 2019, 132, jcs229385. [Google Scholar] [CrossRef]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhász, S.; Künzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Löbrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef]
- Gupta, P.; Saha, B.; Chattopadhyay, S.; Patro, B.S. Pharmacological targeting of differential DNA repair, radio-sensitizes WRN-deficient cancer cells in vitro and in vivo. Biochem. Pharmacol. 2021, 186, 114450. [Google Scholar] [CrossRef]
- Quennet, V.; Beucher, A.; Barton, O.; Takeda, S.; Löbrich, M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011, 39, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Klomp, J.E.; Lee, Y.S.; Goodwin, C.M.; Papke, B.; Klomp, J.A.; Waters, A.M.; Stalnecker, C.A.; DeLiberty, J.M.; Drizyte-Miller, K.; Yang, R.; et al. CHK1 protects oncogenic KRAS-expressing cells from DNA damage and is a target for pancreatic cancer treatment. Cell Rep. 2021, 37, 110060. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Karukonda, P.; Odhiambo, D.; Mowery, Y.M. Pharmacologic inhibition of ataxia telangiectasia and Rad3-related (ATR) in the treatment of head and neck squamous cell carcinoma. Mol. Carcinog. 2022, 61, 225–238. [Google Scholar] [CrossRef]
- Chughtai, A.A.; Pannhausen, J.; Dinger, P.; Wirtz, J.; Knüchel, R.; Gaisa, N.T.; Eble, M.J.; Rose, M. Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR. Biomedicines 2022, 10, 1277. [Google Scholar] [CrossRef]
- Lim, G.; Chang, Y.; Huh, W.-K. Phosphoregulation of Rad51/Rad52 by CDK1 functions as a molecular switch for cell cycle-specific activation of homologous recombination. Sci. Adv. 2020, 6, eaay2669. [Google Scholar] [CrossRef]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Mazina, O.M.; Mazin, A.V. Rad54 protein promotes branch migration of Holliday junctions. Nature 2006, 442, 590–593. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. RAD54 controls access to the invading 3′-OH end after RAD51-mediated DNA strand invasion in homologous recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 2009, 37, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Rossi, M.J.; Mazina, O.M.; Chi, Y.; Moritz, R.L.; Clurman, B.E.; Mazin, A.V. RAD54 N-terminal domain is a DNA sensor that couples ATP hydrolysis with branch migration of Holliday junctions. Nat. Commun. 2018, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. CB 2000, 10, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P. Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat. Rev. Mol. Cell Biol. 2022, 23, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.M.; Recio, M.-J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. CCS 2017, 15, 41. [Google Scholar] [CrossRef]
- Lin, C.-P.; Ban, Y.; Lyu, Y.L.; Liu, L.F. Proteasome-dependent processing of topoisomerase I-DNA adducts into DNA double strand breaks at arrested replication forks. J. Biol. Chem. 2009, 284, 28084–28092. [Google Scholar] [CrossRef]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Sato, K.; Sundaramoorthy, E.; Rajendra, E.; Hattori, H.; Jeyasekharan, A.D.; Ayoub, N.; Schiess, R.; Aebersold, R.; Nishikawa, H.; Sedukhina, A.S.; et al. A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Curr. Biol. CB 2012, 22, 1659–1666. [Google Scholar] [CrossRef]
- Nakamura, K.; Kustatscher, G.; Alabert, C.; Hödl, M.; Forne, I.; Völker-Albert, M.; Satpathy, S.; Beyer, T.E.; Mailand, N.; Choudhary, C.; et al. Proteome dynamics at broken replication forks reveal a distinct ATM-directed repair response suppressing DNA double-strand break ubiquitination. Mol. Cell 2021, 81, 1084–1099.e6. [Google Scholar] [CrossRef] [PubMed]
- Sertic, S.; Mollica, A.; Campus, I.; Roma, S.; Tumini, E.; Aguilera, A.; Muzi-Falconi, M. Coordinated Activity of Y Family TLS Polymerases and EXO1 Protects Non-S Phase Cells from UV-Induced Cytotoxic Lesions. Mol. Cell 2018, 70, 34–47.e4. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Simon, S.E.; Yow, Y.-Y.; Saidur, R.; Tan, K.O. Photoprotective activities of Lignosus rhinocerus in UV-irradiated human keratinocytes. J. Ethnopharmacol. 2022, 299, 115621. [Google Scholar] [CrossRef] [PubMed]
- Trickey, M.; Grimaldi, M.; Yamano, H. The anaphase-promoting complex/cyclosome controls repair and recombination by ubiquitylating Rhp54 in fission yeast. Mol. Cell Biol. 2008, 28, 3905–3916. [Google Scholar] [CrossRef]
- Hadian, K.; Krappmann, D. Signals from the nucleus: Activation of NF-kappaB by cytosolic ATM in the DNA damage response. Sci. Signal. 2011, 4, pe2. [Google Scholar] [CrossRef]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef]
- Gao, S.-S.; Guan, H.; Yan, S.; Hu, S.; Song, M.; Guo, Z.-P.; Xie, D.-F.; Liu, Y.; Liu, X.; Zhang, S.; et al. TIP60 K430 SUMOylation attenuates its interaction with DNA-PKcs in S-phase cells: Facilitating homologous recombination and emerging target for cancer therapy. Sci. Adv. 2020, 6, eaba7822. [Google Scholar] [CrossRef]
- Lange, J.; Yamada, S.; Tischfield, S.E.; Pan, J.; Kim, S.; Zhu, X.; Socci, N.D.; Jasin, M.; Keeney, S. The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 2016, 167, 695–708.e16. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Amé, J.-C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675. [Google Scholar] [CrossRef]
- Yu, Q.; Jiang, Y.; Sun, Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm. Sin. B 2020, 10, 746–765. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Guo, J.; Sun, X.; Gou, W.; Ning, H.; Fang, Z.; Liu, Q.; Hou, W.; Li, Y. Discovery and radiosensitization research of ursolic acid derivatives as SENP1 inhibitors. Eur. J. Med. Chem. 2022, 227, 113918. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.; Warr, N.J.; Shimizu, H.; Watts, F.Z. SUMO modification of Rad22, the Schizosaccharomyces pombe homologue of the recombination protein Rad52. Nucleic Acids Res. 2001, 29, 4179–4186. [Google Scholar] [CrossRef] [PubMed]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar] [CrossRef]
- Zhao, Y.; Hao, S.; Wu, W.; Li, Y.; Hou, K.; Liu, Y.; Cui, W.; Xu, X.; Wang, H. Lysine Crotonylation: An Emerging Player in DNA Damage Response. Biomolecules 2022, 12, 1428. [Google Scholar] [CrossRef]
- Abu-Zhayia, E.R.; Bishara, L.A.; Machour, F.E.; Barisaac, A.S.; Ben-Oz, B.M.; Ayoub, N. CDYL1-dependent decrease in lysine crotonylation at DNA double-strand break sites functionally uncouples transcriptional silencing and repair. Mol. Cell 2022, 82, 1940–1955.e7. [Google Scholar] [CrossRef]
- Yu, H.; Bu, C.; Liu, Y.; Gong, T.; Liu, X.; Liu, S.; Peng, X.; Zhang, W.; Peng, Y.; Yang, J.; et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci. Adv. 2020, 6, eaay4697. [Google Scholar] [CrossRef]
- Kollenstart, L.; de Groot, A.J.L.; Janssen, G.M.C.; Cheng, X.; Vreeken, K.; Martino, F.; Côté, J.; van Veelen, P.A.; van Attikum, H. Gcn5 and Esa1 function as histone crotonyltransferases to regulate crotonylation-dependent transcription. J. Biol. Chem. 2019, 294, 20122–20134. [Google Scholar] [CrossRef]
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer 2022, 21, 99. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Wu, C.; Chang, Y.; Chen, J.; Su, Y.; Li, L.; Chen, Y.; Li, Y.; Wu, J.; Huang, J.; Zhao, F.; et al. USP37 regulates DNA damage response through stabilizing and deubiquitinating BLM. Nucleic Acids Res. 2021, 49, 11224–11240. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Fernando, V.; Letson, J.; Walia, Y.; Zheng, X.; Fackelman, D.; Furuta, S. S-Nitrosylation in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4600. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Filomeni, G. Exploiting S-nitrosylation for cancer therapy: Facts and perspectives. Biochem. J. 2020, 477, 3649–3672. [Google Scholar] [CrossRef] [PubMed]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef]
- Chen, B.; Sun, Y.; Niu, J.; Jarugumilli, G.K.; Wu, X. Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities. Cell Chem. Biol. 2018, 25, 817–831. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, J.M.; Benton, E.R. Shielding effectiveness: A weighted figure of merit for space radiation shielding. Appl. Radiat. Isot. Data Instrum. Methods Use Agric. Ind. Med. 2020, 161, 109141. [Google Scholar] [CrossRef]
- Krukowski, K.; Grue, K.; Becker, M.; Elizarraras, E.; Frias, E.S.; Halvorsen, A.; Koenig-Zanoff, M.; Frattini, V.; Nimmagadda, H.; Feng, X.; et al. The impact of deep space radiation on cognitive performance: From biological sex to biomarkers to countermeasures. Sci. Adv. 2021, 7, eabg6702. [Google Scholar] [CrossRef]
- Deng, M.; Lin, J.; Nowsheen, S.; Liu, T.; Zhao, Y.; Villalta, P.W.; Sicard, D.; Tschumperlin, D.J.; Lee, S.; Kim, J.; et al. Extracellular matrix stiffness determines DNA repair efficiency and cellular sensitivity to genotoxic agents. Sci. Adv. 2020, 6, eabb2630. [Google Scholar] [CrossRef]
- Du, J.; Yin, N.; Xie, T.; Zheng, Y.; Xia, N.; Shang, J.; Chen, F.; Zhang, H.; Yu, J.; Liu, F. Quantitative assessment of HR and NHEJ activities via CRISPR/Cas9-induced oligodeoxynucleotide-mediated DSB repair. DNA Repair 2018, 70, 67–71. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, J.E.; Clarke, D.J.B.; Xie, Z.; Lachmann, A.; Jeon, M.; Chen, K.; Jagodnik, K.M.; Jenkins, S.L.; Kuleshov, M.V.; Wojciechowicz, M.L.; et al. SigCom LINCS: Data and metadata search engine for a million gene expression signatures. Nucleic Acids Res. 2022, 50, W697–W709. [Google Scholar] [CrossRef] [PubMed]
- Boguski, M.S.; Mandl, K.D.; Sukhatme, V.P. Drug discovery. Repurposing with a difference. Science 2009, 324, 1394–1395. [Google Scholar] [CrossRef]
- Kinnings, S.L.; Liu, N.; Buchmeier, N.; Tonge, P.J.; Xie, L.; Bourne, P.E. Drug discovery using chemical systems biology: Repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput. Biol. 2009, 5, e1000423. [Google Scholar] [CrossRef]
- Zhou, X.; Dai, E.; Song, Q.; Ma, X.; Meng, Q.; Jiang, Y.; Jiang, W. In silico drug repositioning based on drug-miRNA associations. Brief. Bioinform. 2020, 21, 498–510. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Wu, C.-K.; Zhang, X.-C.; Yang, Z.-J.; Lu, A.-P.; Hou, T.-J.; Cao, D.-S. Learning to SMILES: BAN-based strategies to improve latent representation learning from molecules. Brief. Bioinform. 2021, 22, bbab327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-C.; Wu, C.-K.; Yang, Z.-J.; Wu, Z.-X.; Yi, J.-C.; Hsieh, C.-Y.; Hou, T.-J.; Cao, D.-S. MG-BERT: Leveraging unsupervised atomic representation learning for molecular property prediction. Brief. Bioinform. 2021, 22, bbab152. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, W.; Ma, J.-L.; Qiu, Y.-L.; Lu, K.; Cao, D.-S.; Wu, C.-K. BioNet: A large-scale and heterogeneous biological network model for interaction prediction with graph convolution. Brief. Bioinform. 2022, 23, bbab491. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhong, Q.; Yang, J.; Duan, Y.; Wang, W.; Wu, C.; He, K. DeepKG: An end-to-end deep learning-based workflow for biomedical knowledge graph extraction, optimization and applications. Bioinforma. Oxf. Engl. 2022, 38, 1477–1479. [Google Scholar] [CrossRef]
- Wang, W.; Yang, X.; Wu, C.; Yang, C. CGINet: Graph convolutional network-based model for identifying chemical-gene interaction in an integrated multi-relational graph. BMC Bioinform. 2020, 21, 544. [Google Scholar] [CrossRef]
- Moore, R.; Puniya, B.L.; Powers, R.; Guda, C.; Bayles, K.W.; Berkowitz, D.B.; Helikar, T. Integrative network analyses of transcriptomics data reveal potential drug targets for acute radiation syndrome. Sci. Rep. 2021, 11, 5585. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef]
- Alok, A.; Agrawala, P.K. Repurposing sodium diclofenac as a radiation countermeasure agent: A cytogenetic study in human peripheral blood lymphocytes. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 856–857, 503220. [Google Scholar] [CrossRef]
- Ren, L.; Xie, D.; Li, P.; Qu, X.; Zhang, X.; Xing, Y.; Zhou, P.; Bo, X.; Zhou, Z.; Wang, S. Radiation protective effects of baclofen predicted by a computational drug repurposing strategy. Pharmacol. Res. 2016, 113, 475–483. [Google Scholar] [CrossRef]
- Dimri, M.; Joshi, J.; Shrivastava, N.; Ghosh, S.; Chakraborti, R.; Indracanti, P.K. Prilocaine hydrochloride protects zebrafish from lethal effects of ionizing radiation: Role of hematopoietic cell expansion. Tokai J. Exp. Clin. Med. 2015, 40, 8–15. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Post-Translational Modification (PTM) | Essential Factor | Description | Application | Reference |
|---|---|---|---|---|
| Phosphorylation | ATM |
|
| [86,87,88,89,90,91,92,93] |
| DNA-PKcs |
|
| [88,94,95,96] | |
| CtIP |
|
| [42,97,98,99] | |
| ATR |
|
| [100,101] | |
| CHK1 |
|
| [101,102,103] | |
| RAD51/52/54 |
|
| [43,70,104,105,106,107] | |
| H2AX |
| / | [108,109] | |
| Ubiquitylation | RNF8 |
|
| [110,111,112] |
| RNF168 |
|
| [47,110,111,113,114,115,116] | |
| REV1 |
|
| [117,118,119,120] | |
| MDM2 |
|
| [89,121,122,123,124] | |
| BRCA1/BARD1 |
|
| [83,125,126,127] | |
| RAD51 |
|
| [128] | |
| Acetylation | Tip60 |
|
| [90,129,130,131] |
| Methylation | BRCA1 |
|
| [132,133,134,135] |
| 53BP1 |
|
| [136,137] | |
| PARylation | PARP1 |
|
| [138,139] |
| Neddylation | NEDD8 |
|
| [140,141,142,143] |
| SUMOylation | / |
|
| [144] |
| MEIIL3 |
|
| [145] | |
| NPM1 |
|
| [146] | |
| Glycosylation | OGT |
|
| [147,148] |
| NEIL3 |
|
| [149,150,151,152] | |
| RUVBL1/2 |
|
| [150,153] | |
| Kcr | CDYL1 |
| / | [154] |
| CBP/P300 |
|
| [155,156,157,158] |
| Era | Condition and Tendency |
|---|---|
| Current |
|
| Future |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, D.; Huang, Q.; Zhou, P. Drug Discovery Targeting Post-Translational Modifications in Response to DNA Damages Induced by Space Radiation. Int. J. Mol. Sci. 2023, 24, 7656. https://doi.org/10.3390/ijms24087656
Xie D, Huang Q, Zhou P. Drug Discovery Targeting Post-Translational Modifications in Response to DNA Damages Induced by Space Radiation. International Journal of Molecular Sciences. 2023; 24(8):7656. https://doi.org/10.3390/ijms24087656
Chicago/Turabian StyleXie, Dafei, Qi Huang, and Pingkun Zhou. 2023. "Drug Discovery Targeting Post-Translational Modifications in Response to DNA Damages Induced by Space Radiation" International Journal of Molecular Sciences 24, no. 8: 7656. https://doi.org/10.3390/ijms24087656