
Specific Activation of T Cells by an ACE2-Based CAR-Like Receptor upon Recognition of SARS-CoV-2 Spike Protein
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ACE2-CAR-Like Design and Expression
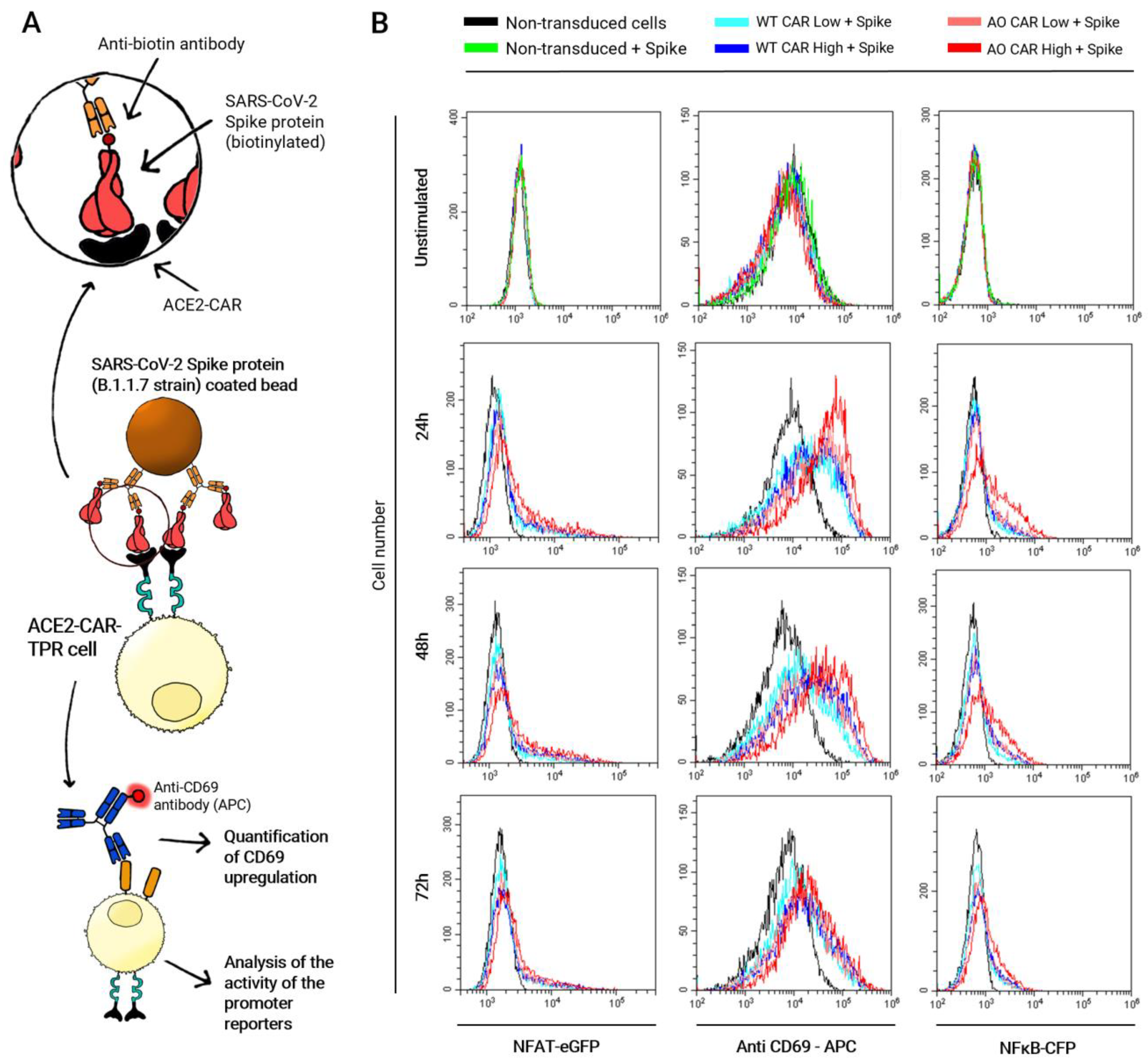
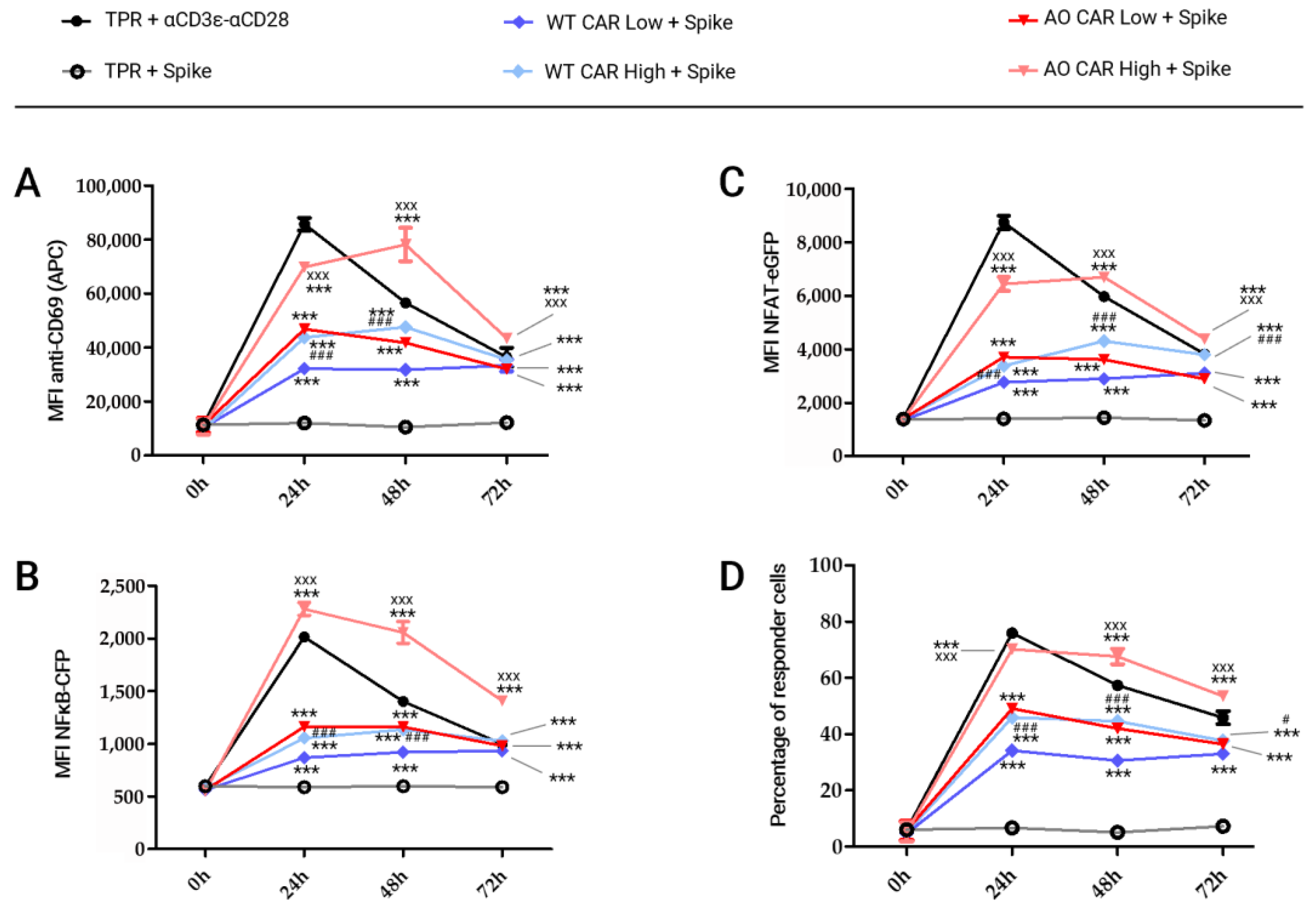
2.2. Kinetic of ACE2-CAR Activation upon Stimulation with Spike Protein-Coated Beads
2.3. Kinetic of ACE2-CAR Activation upon Stimulation with Spike Protein Expressing A549 Cells
3. Discussion
4. Materials and Methods
4.1. CAR Constructs
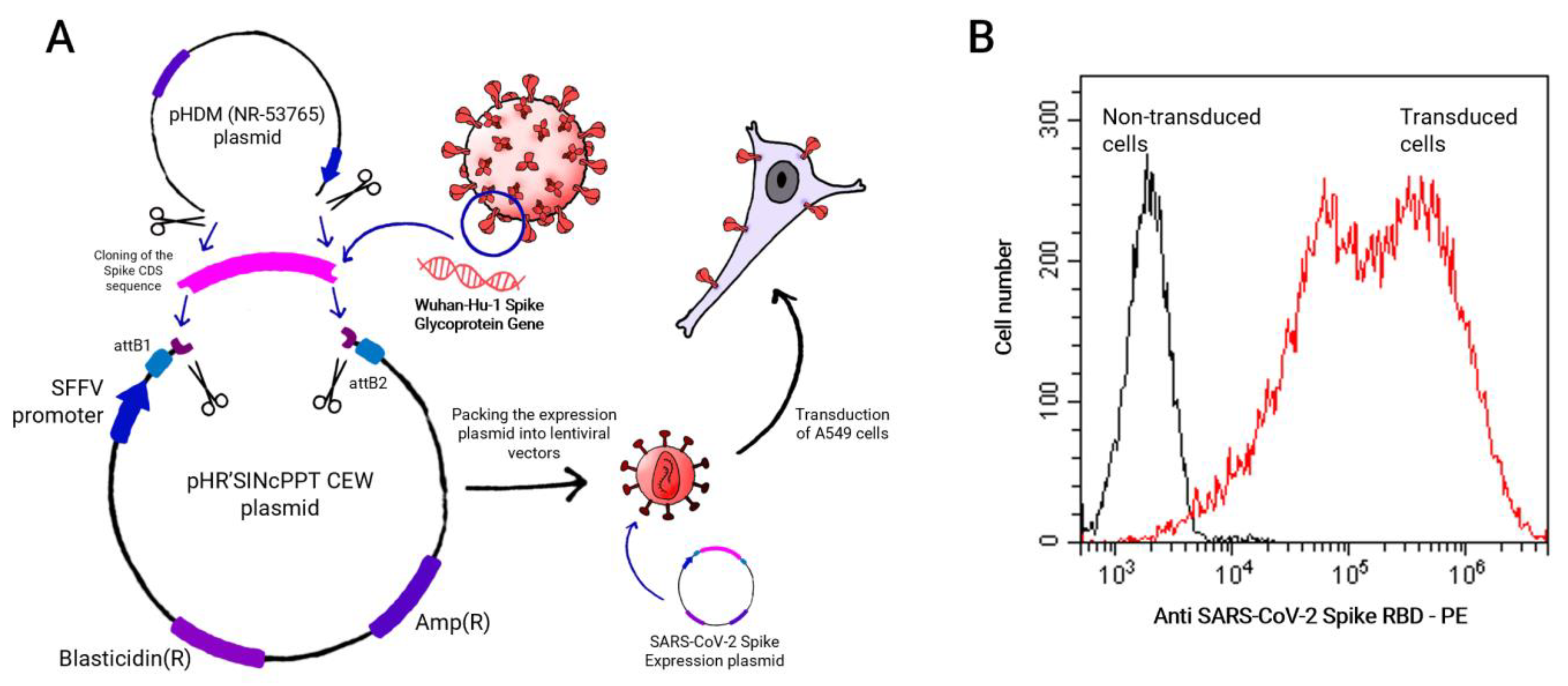
4.2. SARS-CoV-2 Spike Construct
4.3. Cell Lines and Cultures
4.4. Lentiviral Production and Cell Transduction
4.5. Flow Cytometry Analysis of Transduced Cells
4.6. Jurkat-TPR Stimulation Assays
4.7. Statistical Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Banach, M.; Reiner, Ž.; Pirro, M.; Bianconi, V.; Al-Rasadi, K.; Sahebkar, A. Interaction Between Coronavirus S-Protein and Human ACE2: Hints for Exploring Efficient Therapeutic Targets to Treat COVID-19. Angiology 2021, 72, 122–130. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; der Werf, S.V.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Kim, J.M.; Hong, S.P.; Choi, S.Y.; Yang, M.J.; Ju, Y.S.; Kim, Y.T.; Kim, H.M.; Rahman, T.; Chung, M.K.; et al. Nasal ciliated cells are primary targets for SARS-CoV-2 replication in the early stage of COVID-19. J. Clin. Investig. 2021, 131, e148517. [Google Scholar] [CrossRef]
- Wahl, A.; Gralinski, L.; Johnson, C.; Yao, W.; Kovarova, M.; Dinnon, K.; Liu, H.; Madden, V.; Krzystek, H.; De, C.; et al. Acute SARS-CoV-2 Infection is Highly Cytopathic, Elicits a Robust Innate Immune Response and is Efficiently Prevented by EIDD-2801. Res. Sq. 2020, 1–22. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng LF, P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Robinot, R.; Hubert, M.; de Melo, G.D.; Lazarini, F.; Bruel, T.; Smith, N.; Levallois, S.; Larrous, F.; Fernandes, J.; Gellenoncourt, S.; et al. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nat. Commun. 2021, 12, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Laha, S.; Chakraborty, J.; Das, S.; Kanti, S. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect. Genet. Evol. 2020, 85, 104445. [Google Scholar] [CrossRef]
- Paul, D.; Pyne, N.; Paul, S. Mutation profile of SARS-CoV-2 spike protein and identification of potential multiple epitopes within spike protein for vaccine development against SARS-CoV-2. Virus Dis. 2021, 32, 703–726. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Sadoff, J.; Le Gars, M.; Shukarev, G.; Heerwegh, D.; Truyers, C.; de Groot, A.M.; Stoop, J.; Tete, S.; Van Damme, W.; Leroux-Roels, I.; et al. Interim Results of a Phase 1–2a Trial of Ad26.COV2.S COVID-19 Vaccine. N. Engl. J. Med. 2021, 384, 1824–1835. [Google Scholar] [CrossRef]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2022, 29, 344–347. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Zarandi, P.K.; Zinatizadeh, M.; Yousefi, M.H.; Amani, J.; Rezaei, N. Efficacy of mRNA, adenoviral vector, and perfusion protein COVID-19 vaccines. Biomed Pharm. 2022, 146, 112527. [Google Scholar] [CrossRef]
- Galmiche, S.; Luong Nguyen, L.B.; Tartour, E.; de Lamballerie, X.; Wittkop, L.; Loubet, P.; Launay, O. Immunological and clinical efficacy of COVID-19 vaccines in immunocompromised populations: A systematic review. Clin. Microbiol. Infect. 2022, 28, 163–177. [Google Scholar] [CrossRef]
- Shetty, R.; Murugeswari, P.; Chakrabarty, K.; Jayadev, C.; Matalia, H.; Ghosh, A.; Das, D. Stem cell therapy in coronavirus disease 2019: Current evidence and future potential. Cytotherapy 2021, 23, 471–482. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Taha, Y.; Wardle, H.; Evans, A.B.; Hunter, E.R.; Marr, H.; Osborne, W.; Bashton, M.; Smith, D.; Burton-Fanning, S.; Schmid, M.L.; et al. Persistent SARS-CoV-2 infection in patients with secondary antibody deficiency: Successful clearance following combination casirivimab and imdevimab (REGN-COV2) monoclonal antibody therapy. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 85. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Seimetz, D.; Heller, K.; Richter, J. Approval of First CAR-Ts: Have we Solved all Hurdles for ATMPs? Cell Med. 2019, 11, 2155179018822781. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Kazanova, A.; Thurmond, S.; Saragovi, H.U.; Rudd, C.E. Effective chimeric antigen receptor T cells against SARS-CoV-2. Iscience 2021, 24, 103295. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Badeti, S.; Chen, C.-H.; Pinter, A.; Jiang, Q.; Shi, L.; Zhou, R.; Xu, H.; Li, Q.; Gause, W.; et al. CAR-NK Cells Effectively Target the D614 and G614 SARS-CoV-2-infected Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ma, M.T.; Badeti, S.; Chen, C.-H.; Kim, J.; Choudhary, A.; Honnen, B.; Reichman, C.; Calianese, D.; Pinter, A.; Jiang, Q.; et al. CAR-NK Cells Effectively Target SARS-CoV-2-Spike-Expressing Cell Lines In Vitro. Front. Immunol. 2021, 12, 652223. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Ma, Z.; Qian, K.; Li, T.; Zhao, J.; Hu, S. CAR Macrophages for SARS-CoV-2 Immunotherapy. Front. Immunol. 2021, 12, 669103. [Google Scholar] [CrossRef]
- Christodoulou, I.; Rahnama, R.; Ravich, J.W.; Seo, J.; Zolov, S.N.; Marple, A.N.; Markovitz, D.M.; Bonifant, C.L. Glycoprotein Targeted CAR-NK Cells for the Treatment of SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 763460. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Hupperetz, C.; Lah, S.; Kim, H.; Kim, C.H. CAR T Cell Immunotherapy beyond Haematological Malignancy. Immune Netw 2022, 22, e6. [Google Scholar] [CrossRef]
- Zhang, X.; Han, P.; Wang, H.; Xu, Y.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Dai, Q.; Lin, H.; et al. Engineering mesenchymal stromal cells with neutralizing and anti-inflammatory capability against SARS-CoV-2 infection. Mol. Methods Clin. Dev. 2021, 21, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Paidi, R.K.; Jana, M.; Mishra, R.K.; Dutta, D.; Raha, S.; Pahan, K. ACE-2-interacting Domain of SARS-CoV-2 (AIDS) Peptide Suppresses Inflammation to Reduce Fever and Protect Lungs and Heart in Mice: Implications for COVID-19 Therapy. J. Neuroimmune Pharm. 2021, 16, 59–70. [Google Scholar] [CrossRef]
- Min Liu, A.B.; Gao, J. A Phase I/II Study of Universal Off-The-Shelf NKG2D-ACE2 CAR-NK Cells for Therapy of COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04324996 (accessed on 12 March 2023).
- Tsai, T.-I.; Khalili, J.S.; Gilchrist, M.; Waight, A.B.; Cohen, D.; Zhuo, S.; Zhang, Y.; Ding, M.; Zhu, H.; Mak, A.N.-S.; et al. ACE2-Fc fusion protein overcomes viral escape by potently neutralizing SARS-CoV-2 variants of concern. Antivir. Res. 2022, 199, 105271. [Google Scholar] [CrossRef]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, L.; Ullah, I.; Beaudoin-Bussières, G.; Anand, S.P.; Hederman, A.P.; Tolbert, W.D.; Sherburn, R.; Nguyen, D.N.; Marchitto, L.; et al. Engineered ACE2-Fc counters murine lethal SARS-CoV-2 infection through direct neutralization and Fc-effector activities. Sci. Adv. 2022, 8, eabn4188. [Google Scholar] [CrossRef]
- Kabat, E.A. Sequences of Proteins of Immunological Interest, 5th ed.; U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health: Bethesda, MD, USA, 1991. [Google Scholar]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Jutz, S.; Leitner, J.; Schmetterer, K.; Doel-Perez, I.; Majdic, O.; Grabmeier-Pfistershammer, K.; Paster, W.; Huppa, J.B.; Steinberger, P. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: Simultaneous measurement of NF-κB, NFAT and AP-1. J. Immunol. Methods 2016, 430, 10–20. [Google Scholar] [CrossRef]
- Rydzek, J.; Nerreter, T.; Peng, H.; Jutz, S.; Leitner, J.; Steinberger, P.; Einsele, H.; Rader, C.; Hudecek, M. Chimeric Antigen Receptor Library Screening Using a Novel NF-κB/NFAT Reporter Cell Platform. Mol. Ther. 2019, 27, 287–299. [Google Scholar] [CrossRef]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Pühringer, D.; Salzer, B.; Djinović-Carugo, K.; Steinberger, P.; Linhares AD, S.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef] [PubMed]
- Heard, A.; Landmann, J.H.; Hansen, A.R.; Papadopolou, A.; Hsu, Y.-S.; Selli, M.E.; Warrington, J.M.; Lattin, J.; Chang, J.; Ha, H.; et al. Antigen glycosylation regulates efficacy of CAR T cells targeting CD19. Nat. Commun. 2022, 13, 3367. [Google Scholar] [CrossRef]
- Macian, F. Encyclopedia of Molecular Pharmacology; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1119–1126. [Google Scholar]
- Macián, F.; López-Rodríguez, C.; Rao, A. Partners in transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489. [Google Scholar] [CrossRef] [PubMed]
- Macián, F.; García-Rodríguez, C.; Rao, A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000, 19, 4783–4795. [Google Scholar] [CrossRef]
- Lui, I.; Zhou, X.X.; Lim, S.A.; Elledge, S.K.; Solomon, P.; Rettko, N.J.; Zha, B.S.; Kirkemo, L.L.; Gramespacher, J.A.; Liu, J.; et al. Trimeric SARS-CoV-2 Spike interacts with dimeric ACE2 with limited intra-Spike avidity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gavriil, A.; Barisa, M.; Halliwell, E.; Anderson, J. Engineering Solutions for Mitigation of Chimeric Antigen Receptor T-Cell Dysfunction. Cancers 2020, 12, 2326. [Google Scholar] [CrossRef]
- Horndler, L.; Delgado, P.; Abia, D.; Balabanov, I.; Fleta, P.M.; Cornish, G.; Llamas, M.A.; Serrano-Villar, S.; Sánchez-Madrid, F.; Fresno, M.; et al. Flow cytometry multiplexed method for the detection of neutralizing human antibodies to the native SARS-CoV-2 spike protein. EMBO Mol. Med. 2021, 13, e13549. [Google Scholar] [CrossRef]
- Sommers, C.L.; Dejarnette, J.B.; Huang, K.; Lee, J.; El-Khoury, D.; Shores, E.W.; Love, P.E. Function of Cd3ε-Mediated Signals in T Cell Development. J. Exp. Med. 2000, 192, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Brzostek, J.; Sankaran, S.; Wei, Q.; Yap, J.; Tan TY, Y.; Lai, J.; Macary, P.A.; Gascoigne, N.R. Targeting CAR to the Peptide-MHC Complex Reveals Distinct Signaling Compared to that of TCR in a Jurkat T Cell Model. Cancers 2021, 13, 867. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Sharrocks, A.D.; Whitmarsh, A.J. Transcriptional regulation by the MAP kinase signaling cascades. Gene 2003, 320, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Macián, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.T.; May, M.J.; Freedman, B.D. STIM- and Orai-mediated calcium entry controls NF-κB activity and function in lymphocytes. Cell Calcium. 2018, 74, 131–143. [Google Scholar] [CrossRef]
- Bridgeman, J.S.; Ladell, K.; Sheard, V.E.; Miners, K.; Hawkins, R.E.; Price, D.A.; Gilham, D.E. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: Implications for optimization of receptor structure for adoptive cell therapy. Clin. Exp. Immunol. 2014, 175, 258–267. [Google Scholar] [CrossRef]
- Bridgeman, J.S.; Hawkins, R.E.; Bagley, S.; Blaylock, M.; Holland, M.; Gilham, D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010, 184, 6938–6949. [Google Scholar] [CrossRef]
- Tristán-Manzano, M.; Maldonado-Pérez, N.; Justicia-Lirio, P.; Muñoz, P.; Cortijo-Gutiérrez, M.; Pavlovic, K.; Jiménez-Moreno, R.; Nogueras, S.; Carmona, M.; Sánchez-Hernández, S.; et al. Physiological (TCR-like) regulated lentiviral vectors for the generation of improved CAR-T cells. medRxiv 2021. [Google Scholar] [CrossRef]
- Cibrián, D.; Sánchez-Madrid, F. CD69: From activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017, 47, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Poorebrahim, M.; Quiros-Fernandez, I.; Fakhr, E.; Cid-Arregui, A. Generation of CAR-T cells using lentiviral vectors. Methods Cell Biol. 2021, 167, 39–69. [Google Scholar]
- López-Cabrera, M.; Muñoz, E.; Blázquez, M.V.; Ursa, M.A.; Santis, A.G.; Sánchez-Madrid, F. Transcriptional Regulation of the Gene Encoding the Human C-type Lectin Leukocyte Receptor AIM/CD69 and Functional Characterization of Its Tumor Necrosis Factor-α-responsive Elements (∗). J. Biol. Chem. 1995, 270, 21545–21551. [Google Scholar] [CrossRef]
- Zimmerman, M.; Yang, D.; Hu, X.; Liu, F.; Singh, N.; Browning, D.; Ganapathy, V.; Chandler, P.; Choubey, D.; Abrams, S.I.; et al. IFN-γ upregulates survivin and Ifi202 expression to induce survival and proliferation of tumor-specific T cells. PLoS ONE 2010, 5, e14076. [Google Scholar] [CrossRef]
- Hasegawa, A.; Saito, S.; Narimatsu, S.; Nakano, S.; Nagai, M.; Ohnota, H.; Inada, Y.; Morokawa, H.; Nakashima, I.; Morita, D.; et al. Mutated GM-CSF-based CAR-T cells targeting CD116/CD131 complexes exhibit enhanced anti-tumor effects against acute myeloid leukaemia. Clin. Transl. Immunol. 2021, 10, e1282. [Google Scholar] [CrossRef] [PubMed]
- Branella, G.M.; Spencer, H.T. Natural Receptor- and Ligand-Based Chimeric Antigen Receptors: Strategies Using Natural Ligands and Receptors for Targeted Cell Killing. Cells 2021, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Olson, M.L.; Mause ER, V.; Radhakrishnan, S.V.; Brody, J.D.; Rapoport, A.P.; Welm, A.L.; Atanackovic, D.; Luetkens, T. Low-affinity CAR T cells exhibit reduced trogocytosis, preventing rapid antigen loss, and increasing CAR T cell expansion. Leukemia 2022, 36, 1943–1946. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Marquez, P.; Calleja-Cervantes, M.E.; Serrano, G.; Palacios-Berraquero, M.L.; Alignani, D.; San Martin-Uriz, P.; Vilas-Zornoza, A.; Ceballos, C.; Martin-Mallo, A.; Rodríguez-Diaz, S.; et al. CAR Density Influences Antitumoral Efficacy of BCMA CAR-T Cells and Correlates with Clinical Outcome. Blood 2021, 138, 735. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef]
- Worn, A.; Pluckthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Moisini, I.; Geiger, T.L. Identification of a murine CD28 dileucine motif that suppresses single-chain chimeric T-cell receptor expression and function. Blood 2003, 102, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Garson, K.; Li, L.I.; Vanderhyden, B.C. Optimization of lentiviral vector production using polyethylenimine-mediated transfection. Oncol. Lett. 2015, 9, 55–62. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Garcia, P.; Muñoz-Miranda, J.P.; Fernandez-Cisnal, R.; Olvera, L.; Moares, N.; Gabucio, A.; Fernandez-Ponce, C.; Garcia-Cozar, F. Specific Activation of T Cells by an ACE2-Based CAR-Like Receptor upon Recognition of SARS-CoV-2 Spike Protein. Int. J. Mol. Sci. 2023, 24, 7641. https://doi.org/10.3390/ijms24087641
Gonzalez-Garcia P, Muñoz-Miranda JP, Fernandez-Cisnal R, Olvera L, Moares N, Gabucio A, Fernandez-Ponce C, Garcia-Cozar F. Specific Activation of T Cells by an ACE2-Based CAR-Like Receptor upon Recognition of SARS-CoV-2 Spike Protein. International Journal of Molecular Sciences. 2023; 24(8):7641. https://doi.org/10.3390/ijms24087641
Chicago/Turabian StyleGonzalez-Garcia, Pablo, Juan P. Muñoz-Miranda, Ricardo Fernandez-Cisnal, Lucia Olvera, Noelia Moares, Antonio Gabucio, Cecilia Fernandez-Ponce, and Francisco Garcia-Cozar. 2023. "Specific Activation of T Cells by an ACE2-Based CAR-Like Receptor upon Recognition of SARS-CoV-2 Spike Protein" International Journal of Molecular Sciences 24, no. 8: 7641. https://doi.org/10.3390/ijms24087641