

Disruption of Irisin Dimerization by FDA-Approved Drugs: A Computational Repurposing Approach for the Potential Treatment of Lipodystrophy Syndromes



Abstract
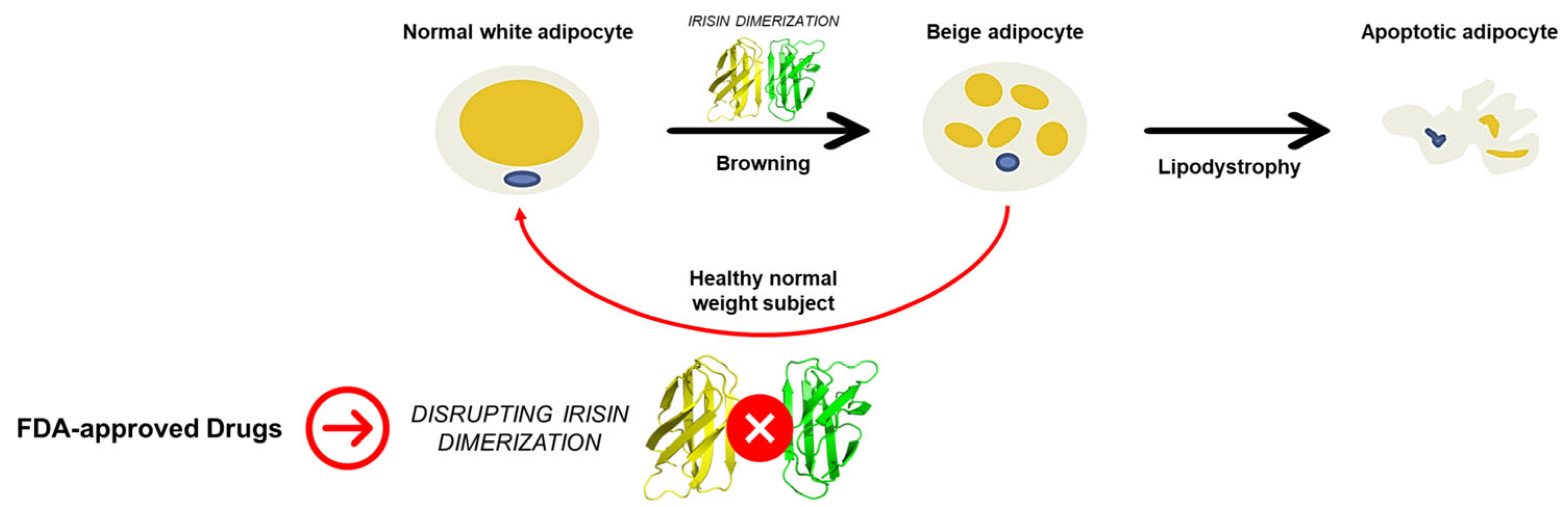
:1. Introduction
2. Results and Discussion
2.1. High-Throughput Docking
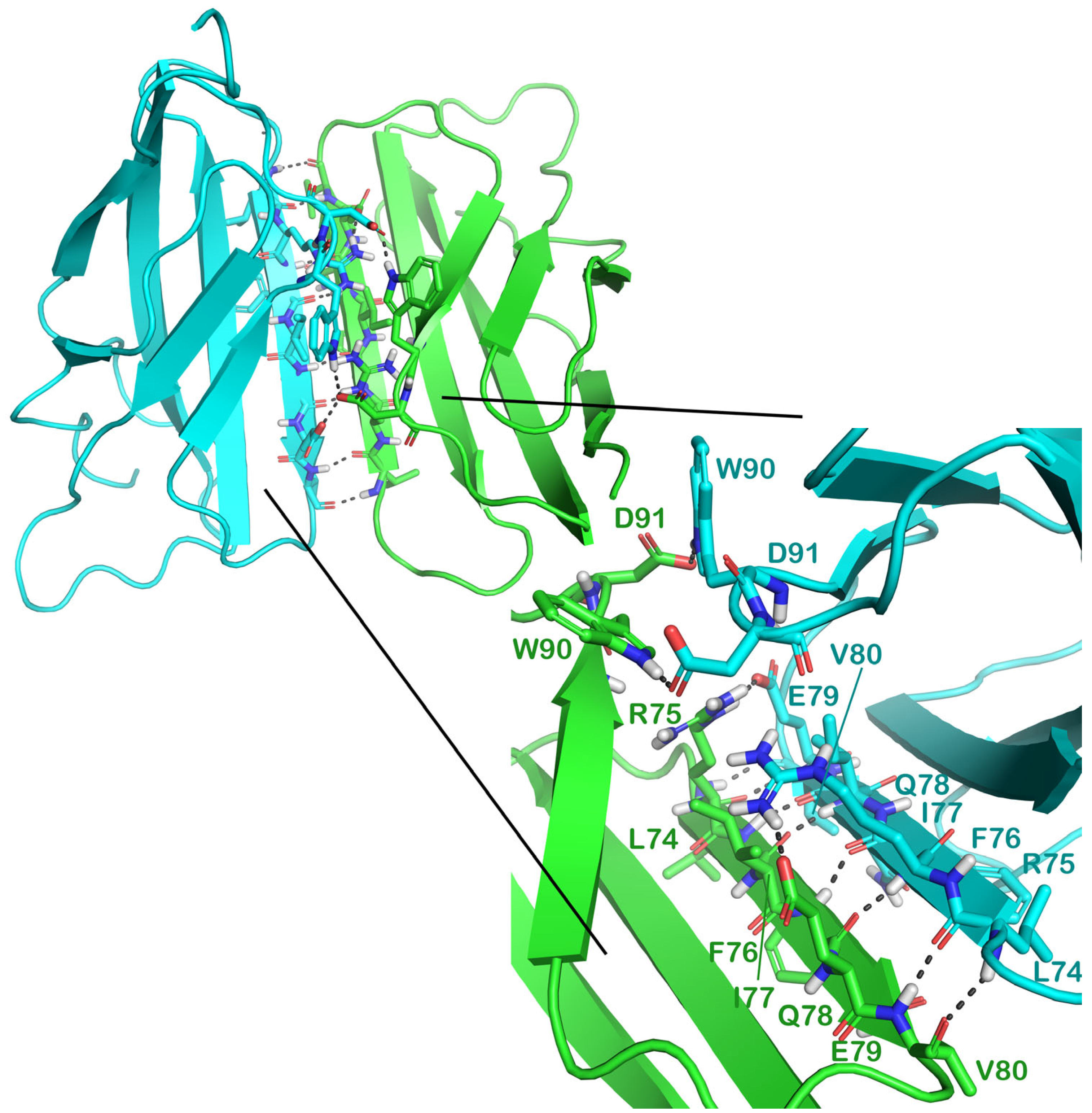
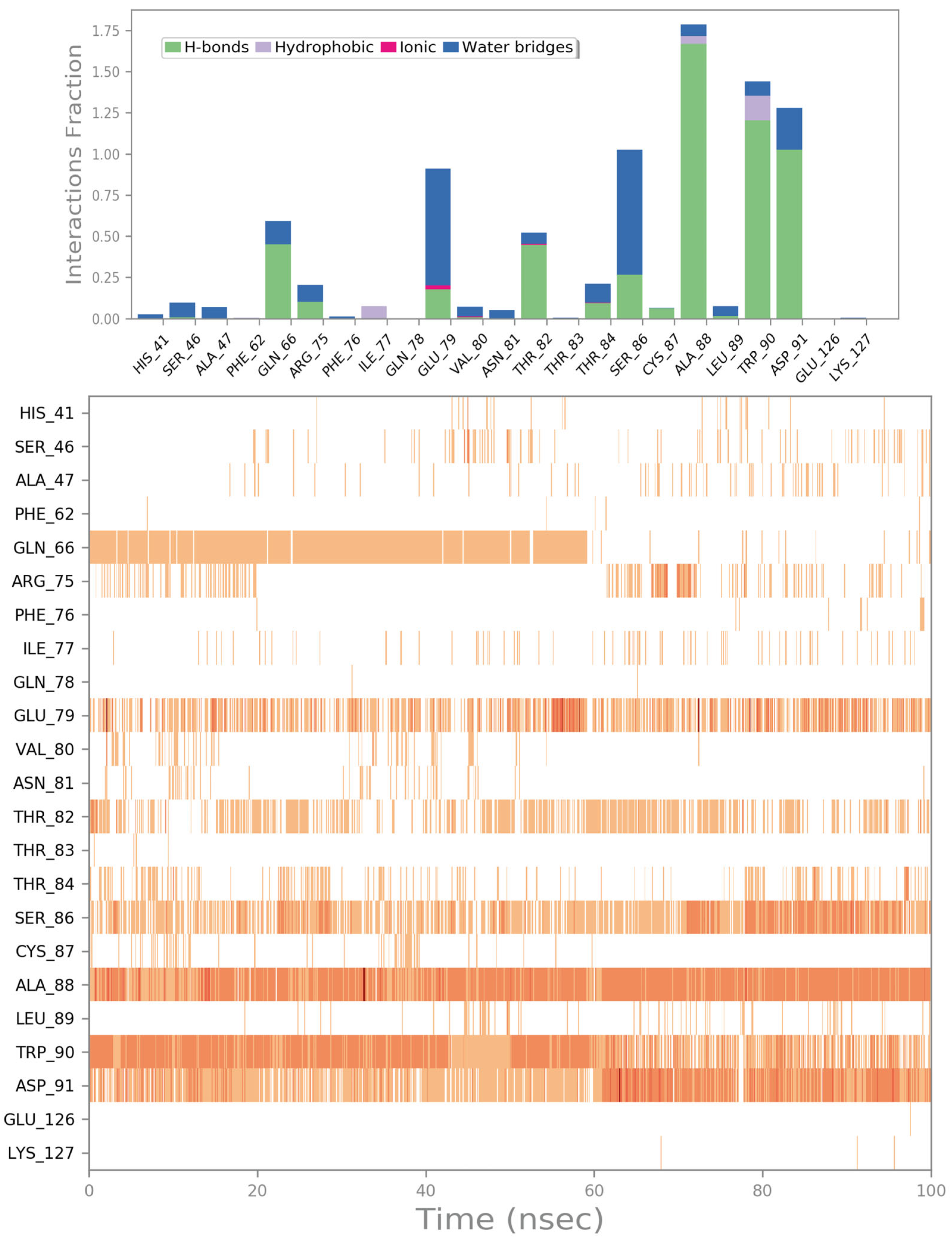
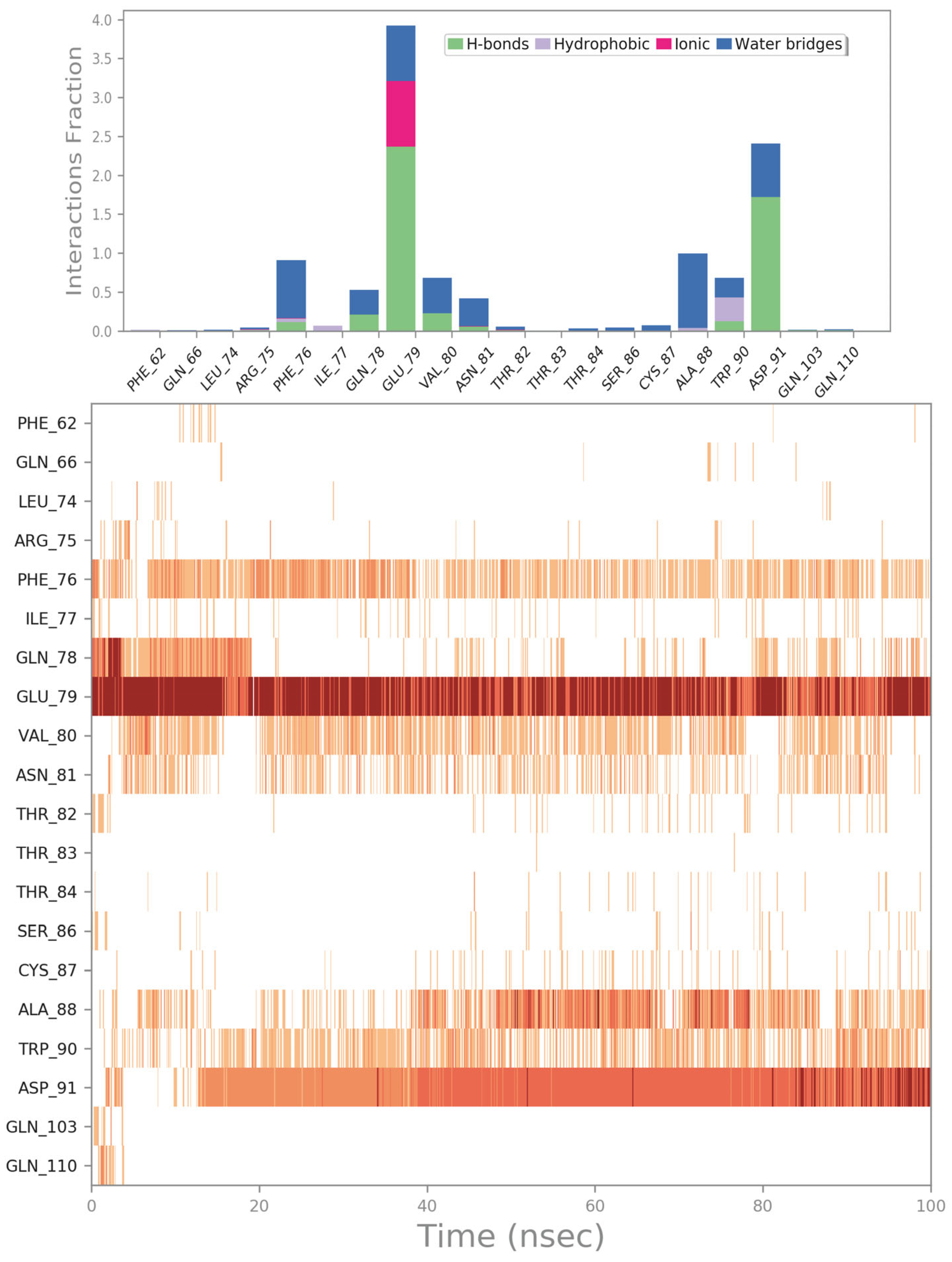
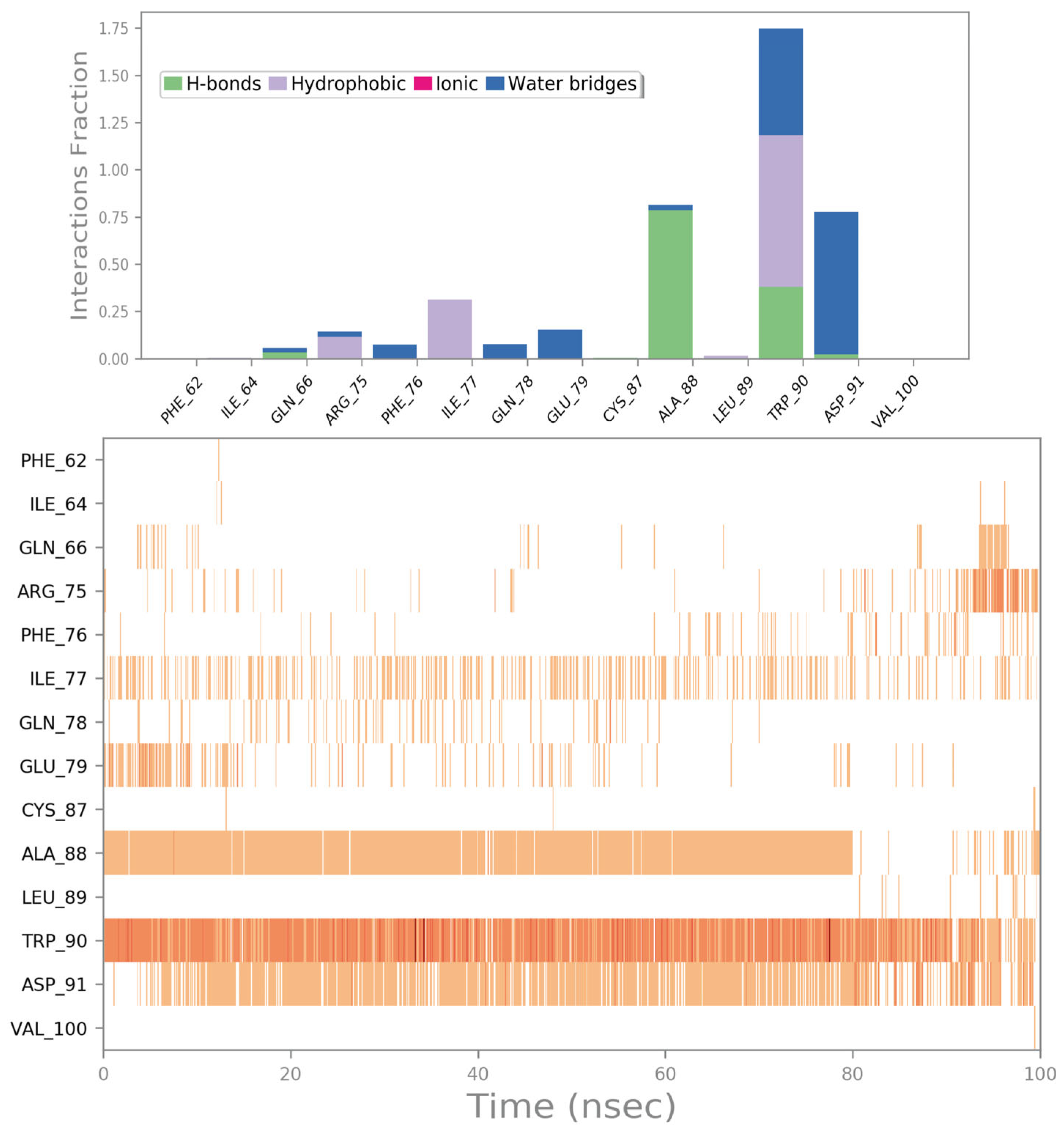
2.2. Molecular Dynamics Simulation
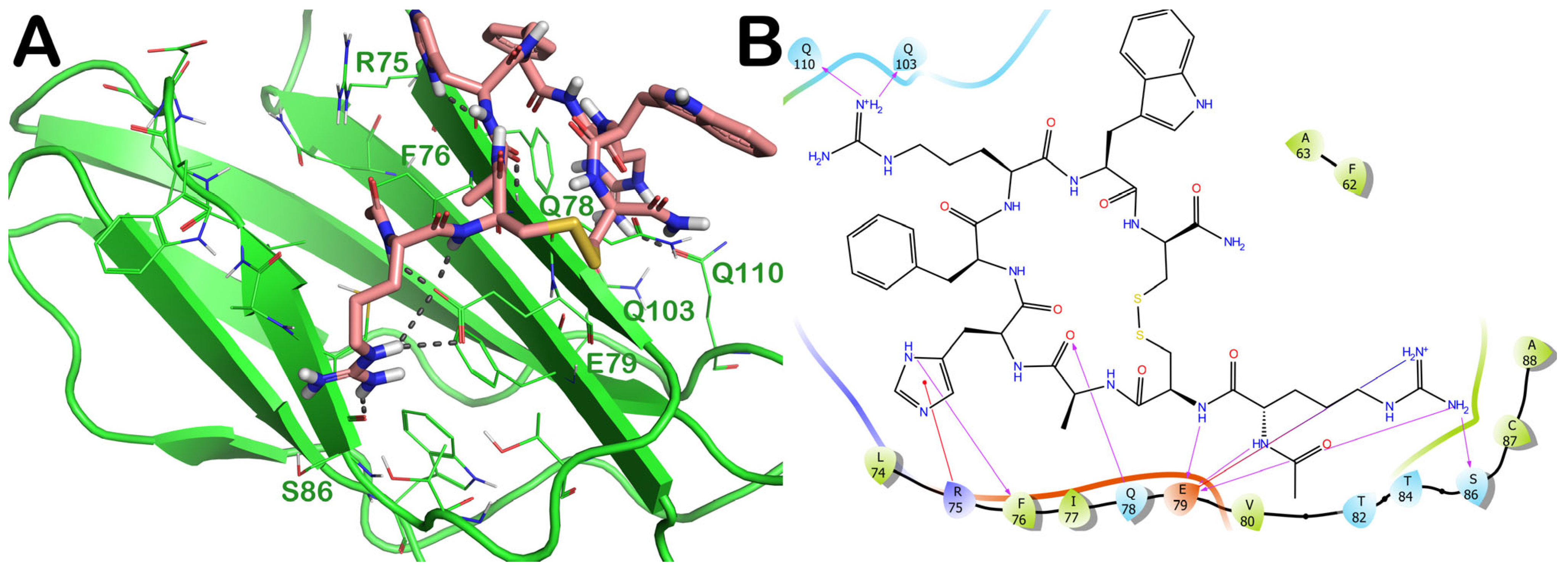
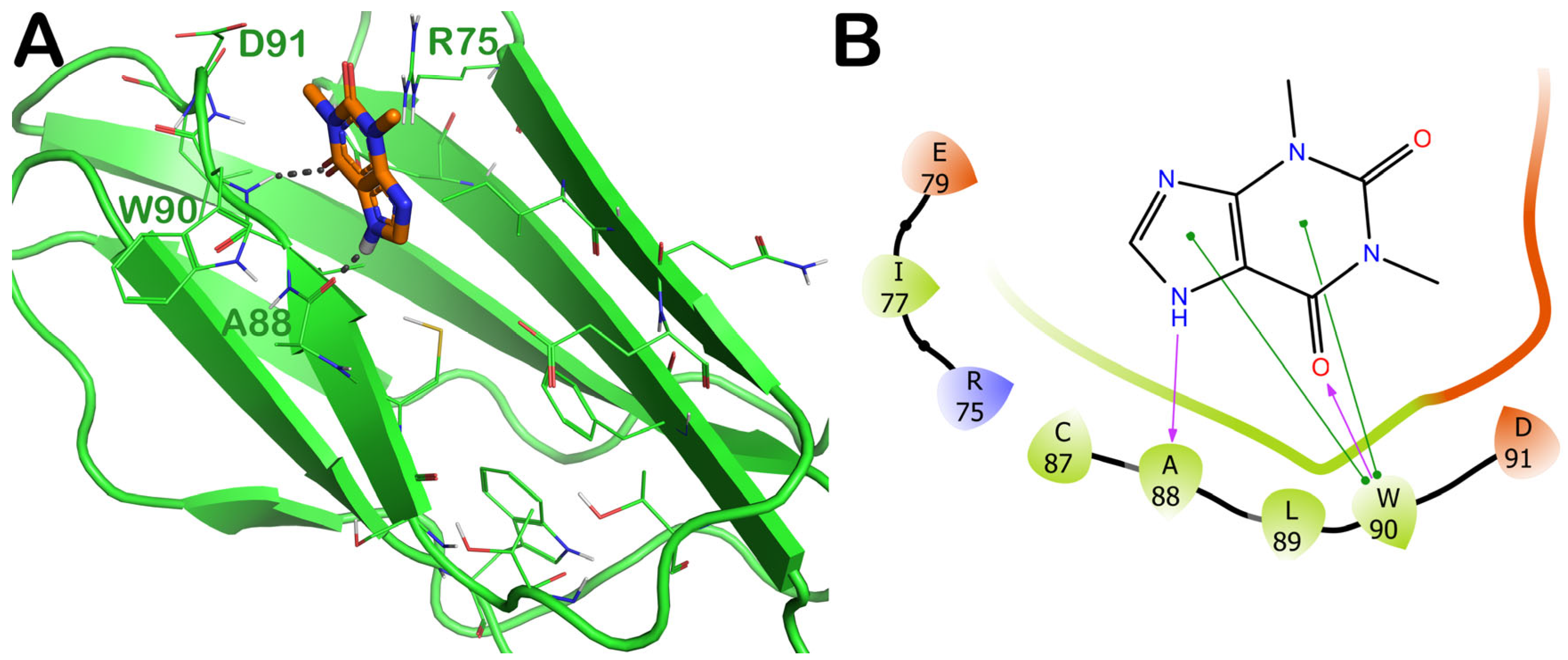
2.3. Potential Hit Compounds Disrupting Irisin Dimerization
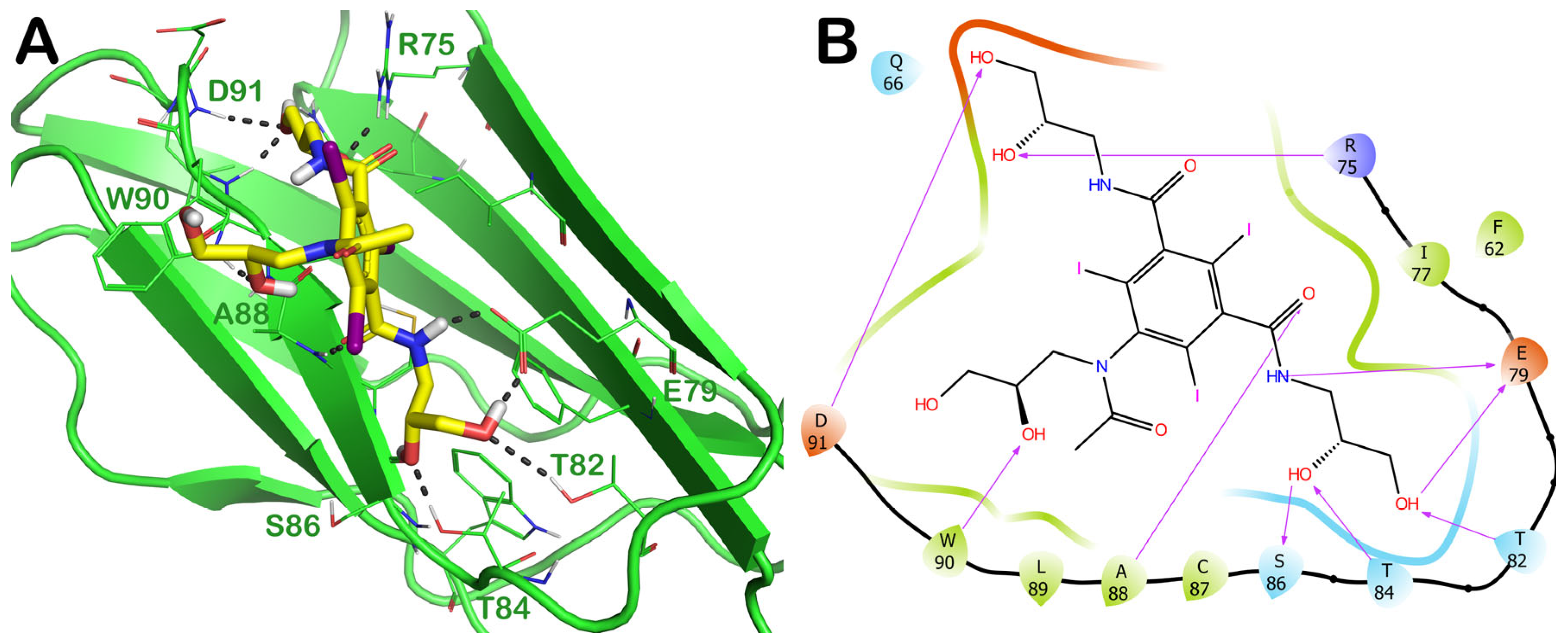
2.3.1. ZINC000003830946 (Iohexol)
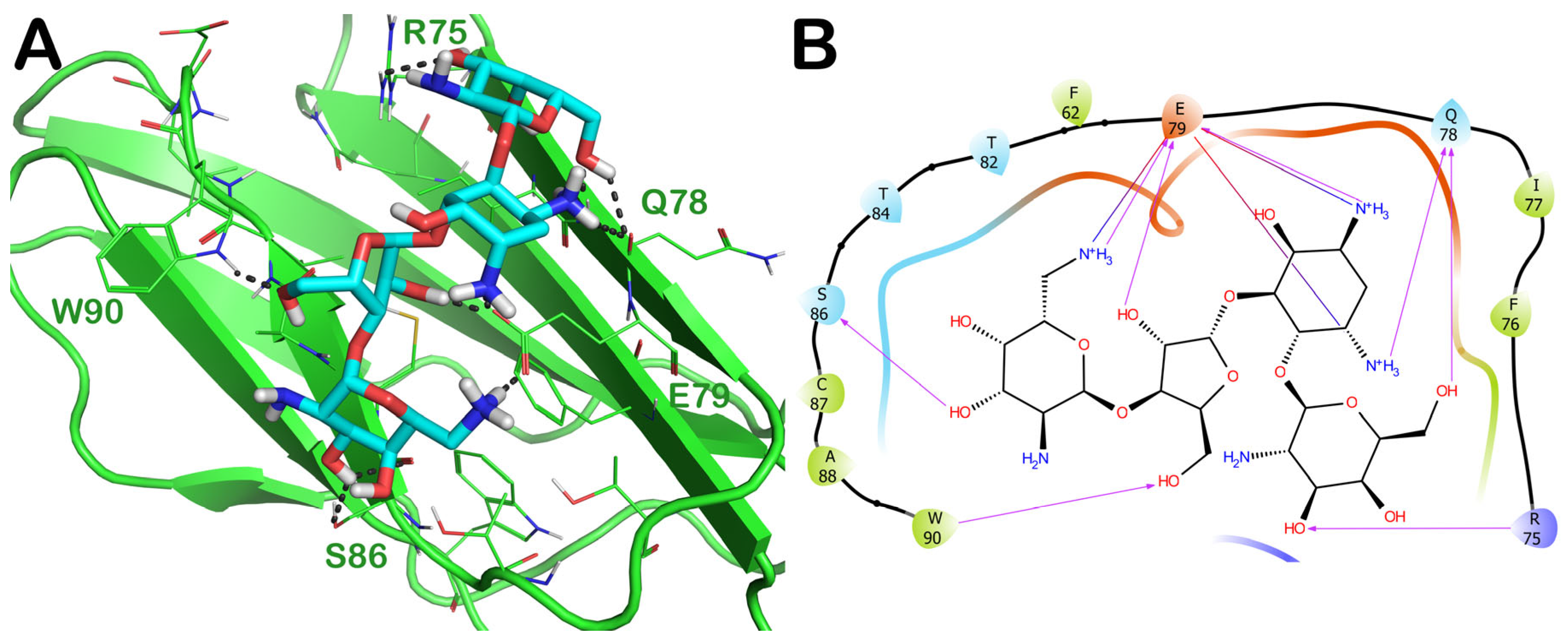
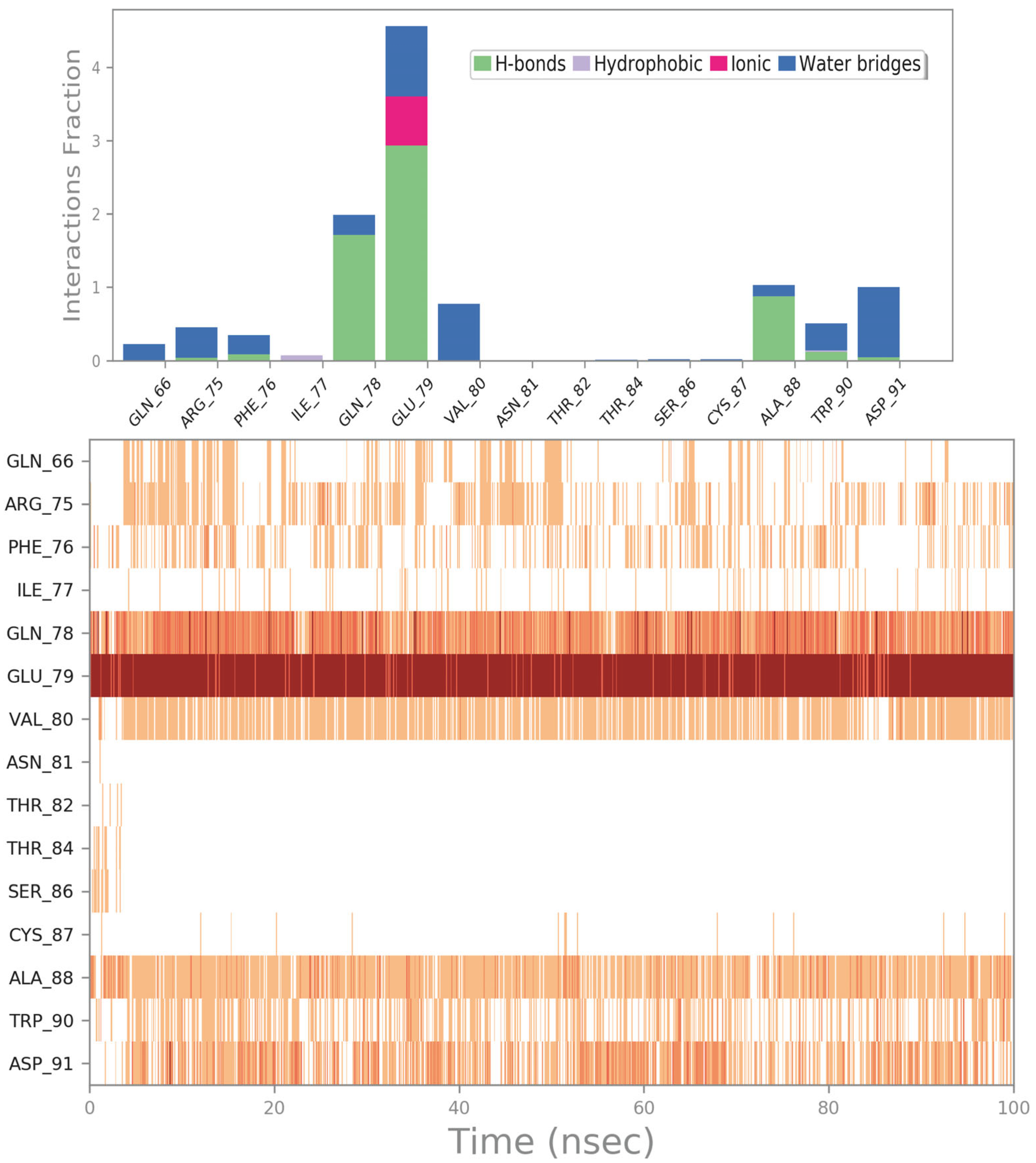
2.3.2. ZINC000060183170 (Paromomycin)
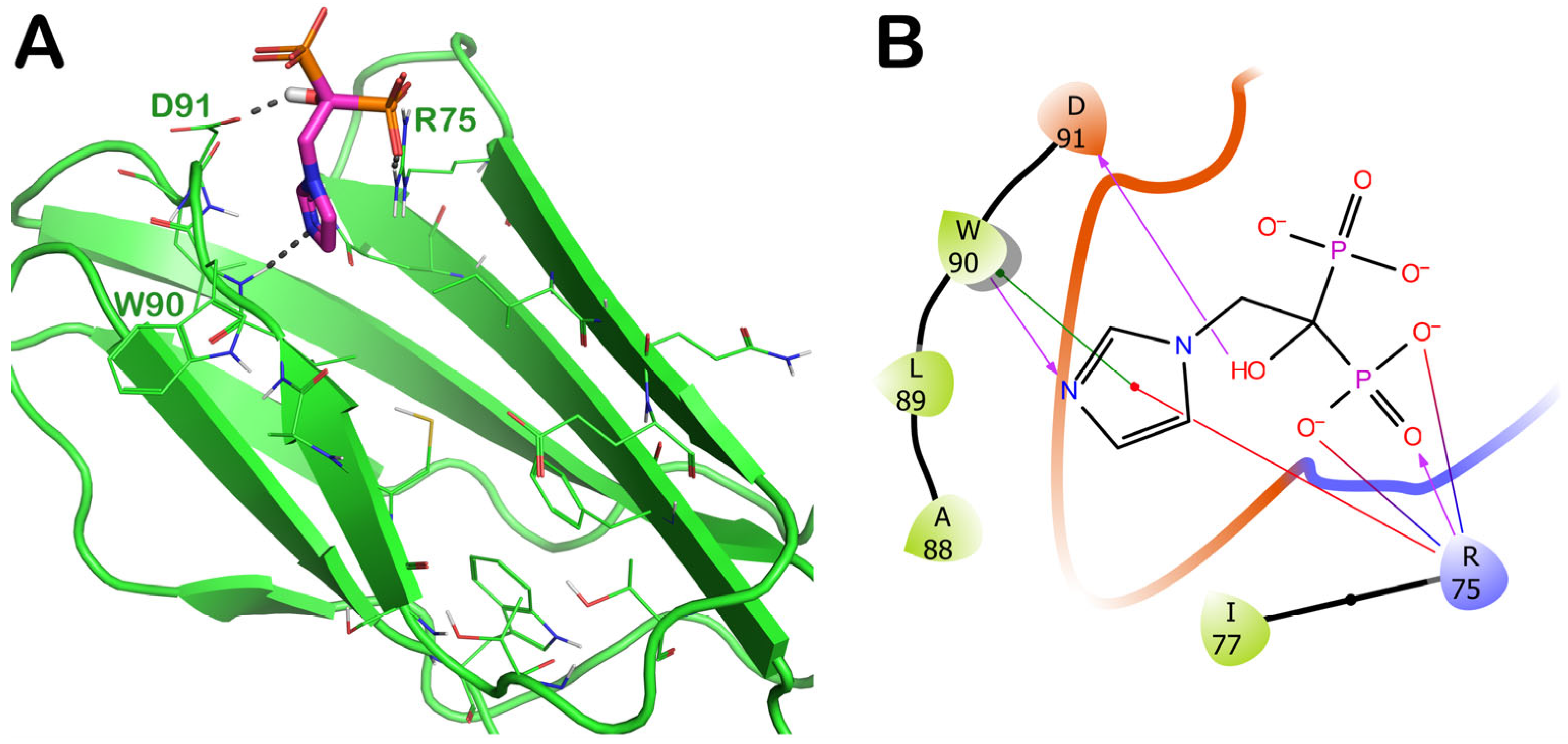
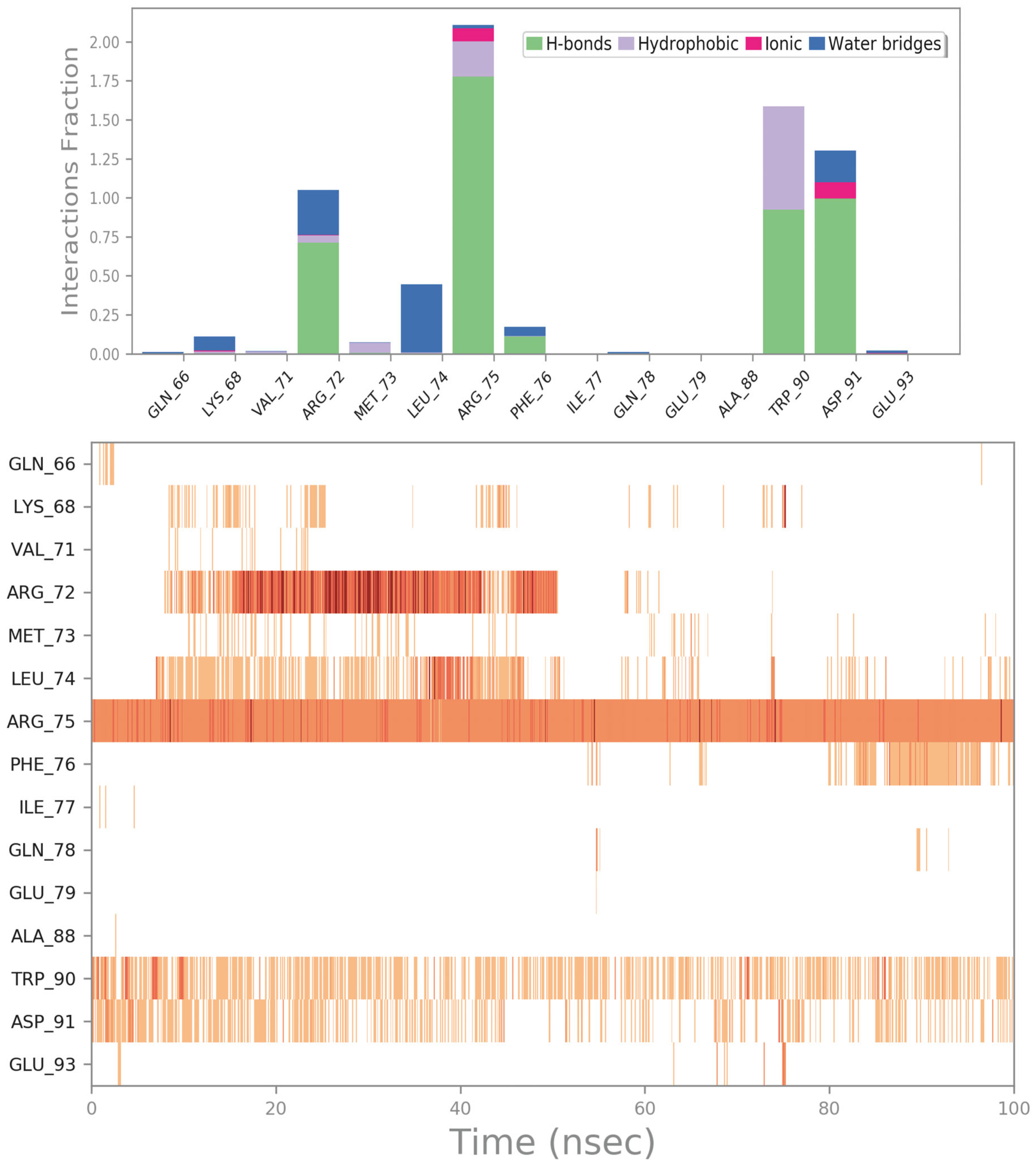
2.3.3. ZINC000003803652 (Zoledronate)
2.3.4. PID: 11993702 (Setmelanotide)
2.3.5. ZINC000018043251 (Theophylline)
2.4. Influence of the Most Promising Drugs against the Irisin Dimerization
3. Materials and Methods
3.1. Computational Details
3.1.1. Protein and World-Approved Drugs Database Preparation
3.1.2. High-Throughput Docking and Ligand Binding Energy Evaluation
3.1.3. Molecular Dynamics
3.1.4. Protein–Protein Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Bensmaine, F.; Benomar, K.; Espiard, S.; Vahe, C.; Le Mapihan, K.; Lion, G.; Lemdani, M.; Chazard, E.; Ernst, O.; Vigouroux, C.; et al. Irisin levels in LMNA-associated partial lipodystrophies. Diabetes Metab. 2019, 45, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Haque, W.A.; Shimomura, I.; Matsuzawa, Y.; Garg, A. Serum adiponectin and leptin levels in patients with lipodystrophies. J. Clin. Endocrinol. Metab. 2002, 87, 2395. [Google Scholar] [CrossRef] [PubMed]
- Safar Zadeh, E.; Lungu, A.O.; Cochran, E.K.; Brown, R.J.; Ghany, M.G.; Heller, T.; Kleiner, D.E.; Gorden, P. The liver diseases of lipodystrophy: The long-term effect of leptin treatment. J. Hepatol. 2013, 59, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Lupsa, B.C.; Sachdev, V.; Lungu, A.O.; Rosing, D.R.; Gorden, P. Cardiomyopathy in congenital and acquired generalized lipodystrophy: A clinical assessment. Medicine 2010, 89, 245–250. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef]
- Diker-Cohen, T.; Cochran, E.; Gorden, P.; Brown, R.J. Partial and generalized lipodystrophy: Comparison of baseline characteristics and response to metreleptin. J. Clin. Endocrinol. Metab. 2015, 100, 1802–1810. [Google Scholar] [CrossRef]
- Lightbourne, M.; Brown, R.J. Genetics of Lipodystrophy. Endocrinol. Metab. Clin. N. Am. 2017, 46, 539–554. [Google Scholar] [CrossRef]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Flori, L.; Testai, L.; Calderone, V. The “irisin system”: From biological roles to pharmacological and nutraceutical perspectives. Life Sci. 2021, 267, 118954. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Chinnam, N.; Ohashi, T.; Shah, R.S.; Erickson, H.P. The structure of irisin reveals a novel intersubunit beta-sheet fibronectin type III (FNIII) dimer: Implications for receptor activation. J. Biol. Chem. 2013, 288, 33738–33744. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Lu, C.; Wang, X.W.; Zhang, J.W.; Song, Y.; Xue, Y.L. Molecular dynamics simulation and steered molecular dynamics simulation on irisin dimers. J. Mol. Model. 2018, 24, 95. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminf. 2020, 12, 46. [Google Scholar] [CrossRef]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the Integration of In Silico Drug Design Methods for Drug Repurposing. Front. Pharmacol. 2017, 8, 298. [Google Scholar] [CrossRef]
- Tuerkova, A.; Zdrazil, B. A ligand-based computational drug repurposing pipeline using KNIME and Programmatic Data Access: Case studies for rare diseases and COVID-19. J. Cheminf. 2020, 12, 71. [Google Scholar] [CrossRef]
- Yalcin-Ozkat, G. Molecular Modeling Strategies of Cancer Multidrug Resistance. Drug Resist. Updat. 2021, 59, 100789. [Google Scholar] [CrossRef]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 2020, 15, 397–401. [Google Scholar] [CrossRef]
- Gazerani, P. Identification of novel analgesics through a drug repurposing strategy. Pain Manag. 2019, 9, 399–415. [Google Scholar] [CrossRef]
- Battah, B.; Chemi, G.; Butini, S.; Campiani, G.; Brogi, S.; Delogu, G.; Gemma, S. A Repurposing Approach for Uncovering the Anti-Tubercular Activity of FDA-Approved Drugs with Potential Multi-Targeting Profiles. Molecules 2019, 24, 4373. [Google Scholar] [CrossRef]
- Aggarwal, S.; Verma, S.S.; Aggarwal, S.; Gupta, S.C. Drug repurposing for breast cancer therapy: Old weapon for new battle. Semin. Cancer Biol. 2021, 68, 8–20. [Google Scholar] [CrossRef]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef]
- Panic, G.; Duthaler, U.; Speich, B.; Keiser, J. Repurposing drugs for the treatment and control of helminth infections. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 185–200. [Google Scholar] [CrossRef]
- Trindade, J.D.S.; Freire-de-Lima, C.G.; Corte-Real, S.; Decote-Ricardo, D.; Freire de Lima, M.E. Drug repurposing for Chagas disease: In Vitro assessment of nimesulide against Trypanosoma cruzi and insights on its mechanisms of action. PLoS ONE 2021, 16, e0258292. [Google Scholar] [CrossRef]
- Cui, C.; Ding, X.; Wang, D.; Chen, L.; Xiao, F.; Xu, T.; Zheng, M.; Luo, X.; Jiang, H.; Chen, K. Drug repurposing against breast cancer by integrating drug-exposure expression profiles and drug-drug links based on graph neural network. Bioinformatics 2021, 37, 2930–2937. [Google Scholar] [CrossRef]
- Roessler, H.I.; Knoers, N.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef]
- Van den Berg, S.; de Visser, S.; Leufkens, H.G.M.; Hollak, C.E.M. Drug Repurposing for Rare Diseases: A Role for Academia. Front. Pharmacol. 2021, 12, 746987. [Google Scholar] [CrossRef]
- Morselli Gysi, D.; do Valle, I.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Ghiassian, S.D.; Patten, J.J.; Davey, R.A.; Loscalzo, J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2025581118. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Sahoo, B.M.; Ravi Kumar, B.V.V.; Sruti, J.; Mahapatra, M.K.; Banik, B.K.; Borah, P. Drug Repurposing Strategy (DRS): Emerging Approach to Identify Potential Therapeutics for Treatment of Novel Coronavirus Infection. Front. Mol. Biosci. 2021, 8, 628144. [Google Scholar] [CrossRef]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.K.; Mukhoty, S.; Kundu, H.; Ghosh, S.; Sen, M.K.; Das, S.; Brogi, S. In silico analysis of RNA-dependent RNA polymerase of the SARS-CoV-2 and therapeutic potential of existing antiviral drugs. Comput. Biol. Med. 2021, 135, 104591. [Google Scholar] [CrossRef] [PubMed]
- Attiq, N.; Arshad, U.; Brogi, S.; Shafiq, N.; Imtiaz, F.; Parveen, S.; Rashid, M.; Noor, N. Exploring the anti-SARS-CoV-2 main protease potential of FDA approved marine drugs using integrated machine learning templates as predictive tools. Int. J. Biol. Macromol. 2022, 220, 1415–1428. [Google Scholar] [CrossRef]
- MacroModel Schrödinger, LLC, New York, NY, USA, 2020. Available online: https://www.schrodinger.com/products/macromodel (accessed on 10 November 2022).
- LigPrep Schrödinger, LLC, New York, NY, USA, 2020. Available online: https://www.schrodinger.com/products/ligprep (accessed on 10 November 2022).
- Brindisi, M.; Brogi, S.; Giovani, S.; Gemma, S.; Lamponi, S.; De Luca, F.; Novellino, E.; Campiani, G.; Docquier, J.D.; Butini, S. Targeting clinically-relevant metallo-beta-lactamases: From high-throughput docking to broad-spectrum inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, S98–S109. [Google Scholar] [CrossRef]
- Brindisi, M.; Brogi, S.; Relitti, N.; Vallone, A.; Butini, S.; Gemma, S.; Novellino, E.; Colotti, G.; Angiulli, G.; Di Chiaro, F.; et al. Structure-based discovery of the first non-covalent inhibitors of Leishmania major tryparedoxin peroxidase by high throughput docking. Sci. Rep. 2015, 5, 9705. [Google Scholar] [CrossRef]
- Zaccagnini, L.; Brogi, S.; Brindisi, M.; Gemma, S.; Chemi, G.; Legname, G.; Campiani, G.; Butini, S. Identification of novel fluorescent probes preventing PrPSc replication in prion diseases. Eur. J. Med. Chem. 2017, 127, 859–873. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Maestro Schrödinger, LLC, New York, NY, USA, 2020. Available online: https://www.schrodinger.com/products/maestro (accessed on 10 November 2022).
- Paolino, M.; Brindisi, M.; Vallone, A.; Butini, S.; Campiani, G.; Nannicini, C.; Giuliani, G.; Anzini, M.; Lamponi, S.; Giorgi, G.; et al. Development of Potent Inhibitors of the Mycobacterium tuberculosis Virulence Factor Zmp1 and Evaluation of Their Effect on Mycobacterial Survival inside Macrophages. Chemmedchem 2018, 13, 422–430. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Prime Schrödinger, LLC, New York, NY, USA, 2020. Available online: https://www.schrodinger.com/products/prime (accessed on 10 November 2022).
- Brindisi, M.; Gemma, S.; Kunjir, S.; Di Cerbo, L.; Brogi, S.; Parapini, S.; D’Alessandro, S.; Taramelli, D.; Habluetzel, A.; Tapanelli, S.; et al. Synthetic spirocyclic endoperoxides: New antimalarial scaffolds. Medchemcomm 2015, 6, 357–362. [Google Scholar] [CrossRef]
- Brogi, S.; Fiorillo, A.; Chemi, G.; Butini, S.; Lalle, M.; Ilari, A.; Gemma, S.; Campiani, G. Structural characterization of Giardia duodenalis thioredoxin reductase (gTrxR) and computational analysis of its interaction with NBDHEX. Eur. J. Med. Chem. 2017, 135, 479–490. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Alfano, G.; Di Cerbo, L.; Brogi, S.; Chemi, G.; Relitti, N.; Brindisi, M.; Lamponi, S.; Novellino, E.; Campiani, G.; et al. Bridged bicyclic 2,3-dioxabicyclo[3.3.1]nonanes as antiplasmodial agents: Synthesis, structure-activity relationships and studies on their biomimetic reaction with Fe(II). Bioorganic Chem. 2019, 89, 103020. [Google Scholar] [CrossRef]
- Schrodinger. Command-Line Only Scripts. Available online: https://www.schrodinger.com/scriptcenter (accessed on 4 February 2023).
- Nickolls, J.; Buck, I.; Garland, M.; Skadron, K. Scalable parallel programming with CUDA. Queue 2008, 6, 40. [Google Scholar] [CrossRef]
- Brogi, S.; Sirous, H.; Calderone, V.; Chemi, G. Amyloid beta fibril disruption by oleuropein aglycone: Long-time molecular dynamics simulation to gain insight into the mechanism of action of this polyphenol from extra virgin olive oil. Food Funct. 2020, 11, 8122–8132. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Sirous, H.; Campiani, G.; Calderone, V.; Brogi, S. Discovery of novel hit compounds as potential HDAC1 inhibitors: The case of ligand- and structure-based virtual screening. Comput. Biol. Med. 2021, 137, 104808. [Google Scholar] [CrossRef]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front. Chem. 2019, 7, 574. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jimenez-Garcia, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Kapoor, N.; Ghorai, S.M.; Khuswaha, P.K.; Bandichhor, R.; Brogi, S. Butein as a potential binder of human ACE2 receptor for interfering with SARS-CoV-2 entry: A computer-aided analysis. J. Mol. Model. 2022, 28, 270. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipodystrophy Type | Mutation/Alteration | Gene Involved | Protein Involved |
|---|---|---|---|
| Congenital generalized lipodystrophy | Recessive mutations | AGPAT2 | Adipocytes growth (glycerophospholipids and triacylglycerols) |
| BSCL2 | Seipin (endoplasmic reticulum, development of lipid droplets) | ||
| CAV1 | Caveolin-1 (essential for the normal transport, processing, and storage of fats) | ||
| PTRF | Caveolin-3 (caveolar formation) | ||
| Familial partial lipodystrophy | Autosomal dominant mutations | LMNA | Lamin A/C (stability and strength to cells) |
| PPARG | PPARγ (key role in lipid and glucose metabolism) | ||
| PLIN1 | Perilipin-1 (coats lipid storage droplets) | ||
| AKT2 | AKT Kinase (metabolism, proliferation, growth, and angiogenesis) | ||
| Recessive mutations | CIDEC | CIDE-3 (lipolysis restriction and favored storage) | |
| LIPE | Lipase E, Hormone Sensitive (mobilization of triglycerides from adipose tissue) | ||
| Acquired lipodystrophies | Autoimmune destruction of adipocytes | - | - |
| Acquired partial lipodystrophy | Mutations | LMNB | Lamin-like protein (maintain integrity of nuclear structures in response to mechanical stress) |
| No | Compound | Status | XP Score (kcal/Mol) | SP Score (kcal/Mol) | ΔGbind (kcal/Mol) | Binding Stability a |
|---|---|---|---|---|---|---|
| 1 | ZINC000003830946 PID: 3730 Iohexol (Omnipaque) | FDA-approved | −7.70 | −5.52 | −61.47 | yes |
| 2 | PID: 11993702 Setmelanotide (Imcivree) | FDA-approved EMA-approved | −6.10 | −7.24 | −56.87 | yes |
| 3 | ZINC000008551963 PID: 148197 Diquafosol (Diquas) | Experimental FDA UNII 7828VC80FJ PMDA-approved | −8.17 | −5.02 | −54.91 | yes |
| 4 | ZINC000008214585 PID: 3037209 Isepamicin | Experimental FDA UNII G7K224460P PMDA-approved | −7.94 | −5.70 | −51.72 | no |
| 5 | ZINC000003830947 PID: 65492 Iopamidol | FDA-approved | −5.94 | −5.84 | −51.49 | no |
| 6 | ZINC000060183170 PID: 165580 Paromomycin (Humatin) | FDA-approved | −7.23 | −6.18 | −50.14 | yes |
| 7 | ZINC000085504689 PID: 5281771 Echinacoside | Experimental FDA UNII I04O1DT48T | −5.68 | −5.36 | −48.36 | no |
| 8 | ZINC000085552699 PID: 5486699 Troxerutin | Experimental FDA UNII 7Y4N11PXO8 | −7.45 | −5.13 | −46.18 | no |
| 9 | ZINC000003830273 PID: 2327 Benserazide (component of Prolopa) | FDA-approved | −6.71 | −6.37 | −44.16 | no |
| 10 | ZINC000238809356 PID: 13943297 Astragaloside IV | Experimental FDA UNII 1J6XA9YCFV | −6.46 | −5.09 | −43.39 | no |
| 11 | ZINC000003946372 PID: 9808998 Edotecarin | Experimental FDA UNII 1V8X590XDP | −6.00 | −5.91 | −43.32 | no |
| 12 | ZINC000003788703 PID: 444020 Voglibose (Basen) | PMDA-approved Experimental FDA UNII S77P977AG8 | −7.46 | −5.42 | −42.11 | yes |
| 13 | ZINC000008143568 PID: 10621 Hesperidin | Experimental FDA UNII E750O06Y6O | −6.58 | −5.03 | −41.69 | no |
| 14 | ZINC000008214483 PID: 37768 Amikacin (Arikayce) | FDA-approved EMA-approved | −7.11 | −5.25 | −41.25 | no |
| 15 | ZINC000049538629 PID: 6451084 Salvianolic acid B | Experimental FDA UNII C1GQ844199 | −5.10 | −6.04 | −40.67 | yes |
| 16 | ZINC000003977952 PID: 11333 Lactulose (Generlac) | FDA-approved | −6.38 | −5.85 | −35.66 | no |
| 17 | ZINC000004096261 PID: 638678 Protirelin (Thyrel TRH) | FDA-approved | −5.63 | −5.34 | −34.88 | no |
| 18 | ZINC000018043251 PID: 2153 Theophylline (Quibron-T) | FDA-approved | −5.17 | −5.55 | −33.25 | yes |
| 19 | ZINC000003938642 PID: 451417 Thymopentin | Experimental FDA UNII O3Y80ZF13F | −5.32 | −5.92 | −32.73 | no |
| 20 | ZINC000003803652 PID: 68740 Zoledronate (Zometa) | FDA-approved EMA-approved | −6.33 | −5.53 | −32.38 | yes |
| 21 | ZINC000085537042 PID: 41774 Acarbose (Glucobay; Precose) | FDA-approved | −6.65 | −5.11 | −31.46 | no |
| 22 | ZINC000000057624 PID: 439260 Noradrenaline (Levophed) | FDA-approved | −5.03 | −5.49 | −30.59 | no |
| 23 | ZINC000002040854 PID: 439224 Carnosine (Sevitin) | Experimental FDA UNII 8HO6PVN24W PMDA-approved | −6.04 | −5.25 | −29.54 | no |
| Entry | ΔGvdw a (kcal/mol) | ΔGcoul b (kcal/mol) | ΔGHbond c (kcal/mol) | ΔGLipo d (kcal/mol) | ΔGPack e (kcal/mol) | ΔGSolGB f (kcal/mol) | ΔGbind g (kcal/mol) |
|---|---|---|---|---|---|---|---|
| ZINC000003830946 (Iohexol) | −33.84 | −37.12 | −4.39 | −10.45 | −0.11 | 23.05 | −60.71 |
| ZINC000060183170 (Paromomycin) | −32.14 | −93.71 | −5.52 | −12.38 | 0.00 | 89.45 | −49.13 |
| ZINC000003803652 (Zoledronate) | −11.67 | 42.11 | −3.58 | −2.78 | −3.09 | −31.73 | −29.42 |
| PID: 11993702 (Setmelanotide) | −54.67 | −113.82 | −6.28 | −15.23 | −5.15 | 98.12 | −62.41 |
| ZINC000018043251 (Theophylline) | −19.56 | −18.92 | −1.11 | −5.19 | −7.69 | 13.18 | −35.29 |
| Complex | Clusters | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
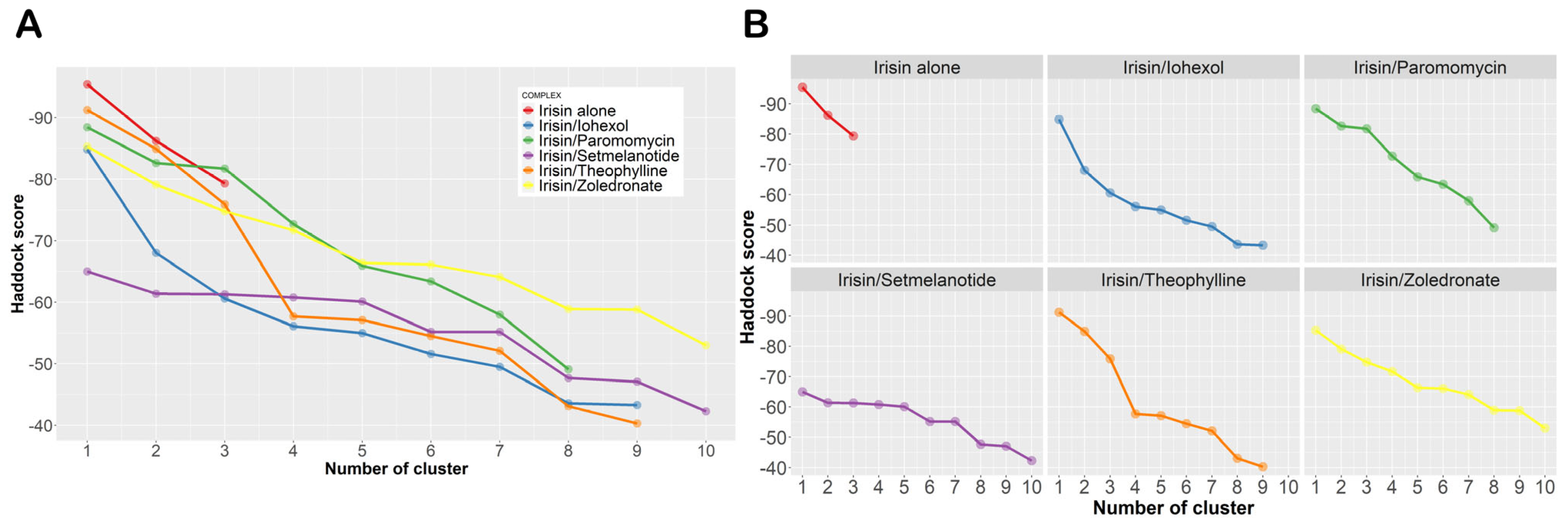
| Irisin alone | −95.4 ± 3.5 (87%) | −86.2 ± 5.4 (8%) | −79.3 ± 2.7 (5%) | |||||||
| Irisin/iohexol | −84.8 ± 6.5 (31%) | −68.0 ± 4.8 (28%) | 60.6 ± 1.0 (11%) | −56.1 ± 5.5 (3%) | −55.0 ± 8.0 (5%) | −51.6 ± 3.9 (7%) | −49.5 ± 2.7 (6%) | −43.6 ± 11.9 (3%) | −43.3 ± 3.1 (3%) | |
| Irisin/paromomycin | −88.4 ± 7.0 (14%) | −82.6 ± 3.0 (52%) | −81.7 ± 1.4 (11%) | −72.7 ± 11.2 (10%) | −65.9 ± 8.4 (5%) | −63.4 ± 3.2 (3%) | −58.0 ± 8.2 (3%) | −49.1 ± 11.3 (2%) | ||
| Irisin/zoledronate | −85.3 ± 3.1 (41%) | −79.1 ± 5.7 (19%) | −74.8 ± 8.5 (8%) | −71.7 ± 6.3 (5%) | −66.4 ± 7.8 (6%) | −66.1 ± 6.4 (3%) | −64.1 ± 11.6 (4%) | −58.9 ± 8.7 (4%) | −58.8 ± 8.8 (7%) | −53.0 ± 5.0 (3%) |
| Irisin/setmelanotide | −65.0 ± 3.7 (22%) | −61.4 ± 2.1 (24%) | −61.3 ± 9.2 (3%) | −60.8 ± 2.0 (11%) | −60.1 ± 7.4 (5%) | −55.2 ± 3.3 (14%) | −55.2 ± 9.5 (8%) | −47.7 ± 7.4 (5%) | −47.1 ± 1.4 (4%) | −42.3 ± 3.4 (4%) |
| Irisin/theophylline | −91.2 ± 3.0 (11%) | −84.9 ± 5.4 (50%) | −75.9 ± 3.1 (9%) | −57.7 ± 4.5 (6%) | −57.1 ± 2.9 (8%) | −54.5 ± 7.1 (3%) | −52.1 ± 4.0 (6%) | −43.1 ± 1.0 (4%) | −40.3 ± 6.9 (3%) | |
| Drugs | Marketed Indication | Note |
|---|---|---|
 Iohexol | Diagnostic tool used for X-ray imaging | Blocks X-rays as they pass through the body |
 Paromomycin | Treatment of acute and chronic intestinal amebiasis, cutaneous and mucocutaneous leishmaniasis, and as an adjuvant for the management of hepatic coma | Aminoglycoside antibiotic that inhibits protein synthesis by binding to 16S ribosomal RNA |
 Zoledronic acid | Treatment of malignancy-associated hypercalcemia, multiple myeloma, and bone metastasis from solid tumors | Bisphosphonate; acts on bone tissue by inhibiting the bone resorption process mediated by osteoclasts |
 Setmelanotide | Treatment for patients with proopiomelanocortin, proprotein subtilisin/kexin type 1, or leptin deficiencies. Used for treating genetic obesity caused by the mentioned rare single-gene mutations | Melanocortin 4 receptor agonist |
 Theophylline | Treatment of the symptoms and reversible airflow obstruction associated with chronic asthma and other chronic lung diseases, such as emphysema and chronic bronchitis | Competitively inhibits type III and type IV phosphodiesterase (PDE) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flori, L.; Brogi, S.; Sirous, H.; Calderone, V. Disruption of Irisin Dimerization by FDA-Approved Drugs: A Computational Repurposing Approach for the Potential Treatment of Lipodystrophy Syndromes. Int. J. Mol. Sci. 2023, 24, 7578. https://doi.org/10.3390/ijms24087578
Flori L, Brogi S, Sirous H, Calderone V. Disruption of Irisin Dimerization by FDA-Approved Drugs: A Computational Repurposing Approach for the Potential Treatment of Lipodystrophy Syndromes. International Journal of Molecular Sciences. 2023; 24(8):7578. https://doi.org/10.3390/ijms24087578
Chicago/Turabian StyleFlori, Lorenzo, Simone Brogi, Hajar Sirous, and Vincenzo Calderone. 2023. "Disruption of Irisin Dimerization by FDA-Approved Drugs: A Computational Repurposing Approach for the Potential Treatment of Lipodystrophy Syndromes" International Journal of Molecular Sciences 24, no. 8: 7578. https://doi.org/10.3390/ijms24087578