
In Silico Design of a Chimeric Humanized L-asparaginase

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
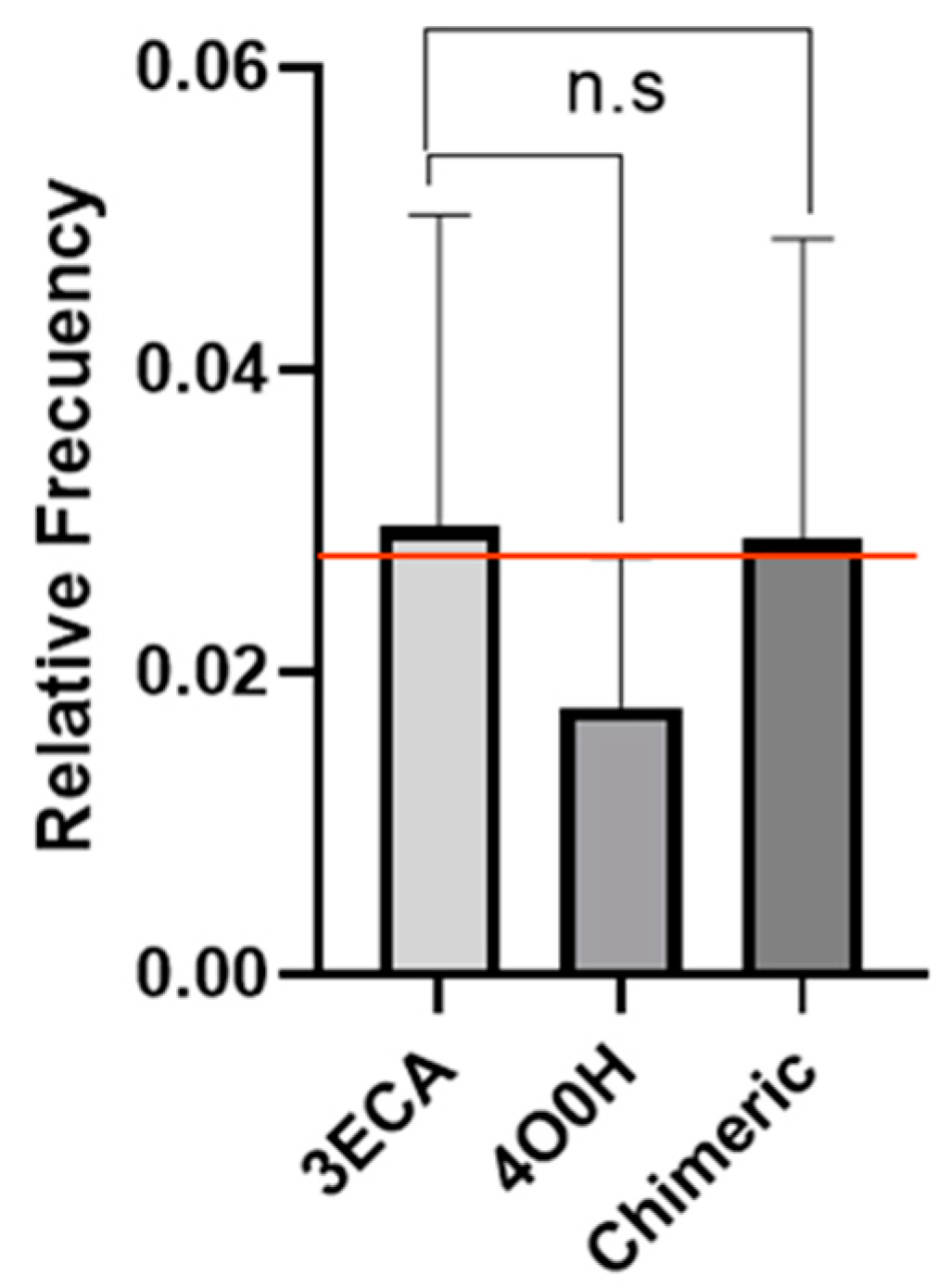
2.1. Determination of Epitope Density
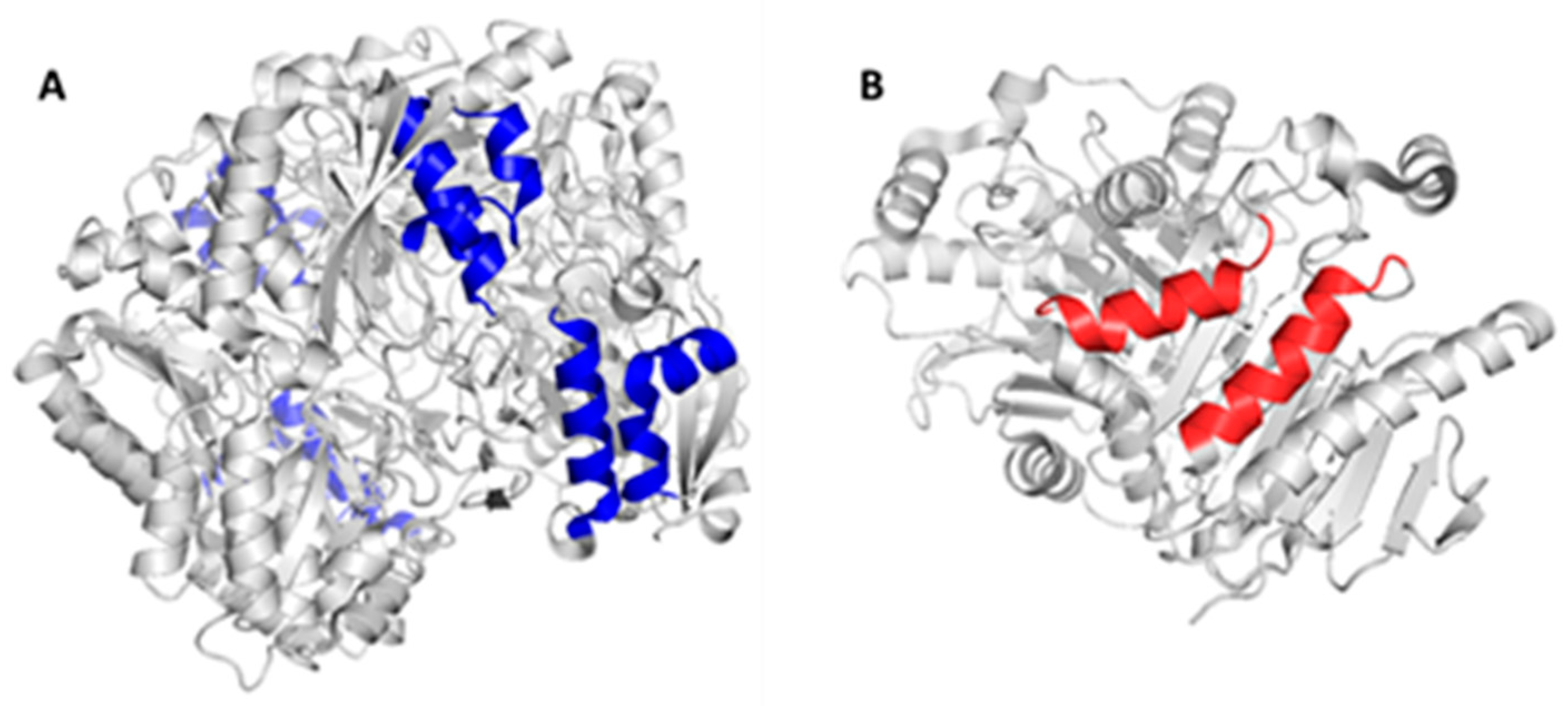
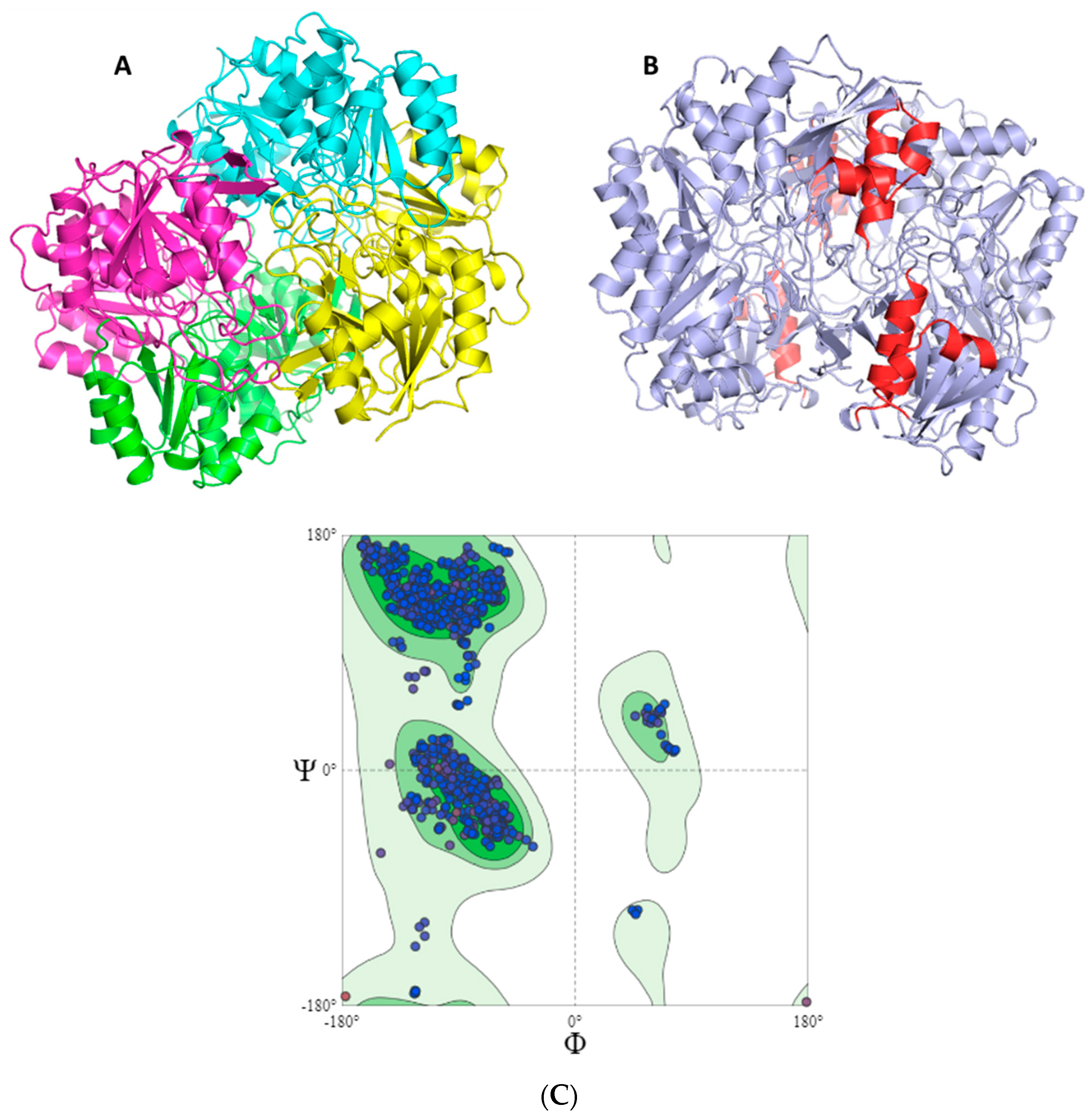
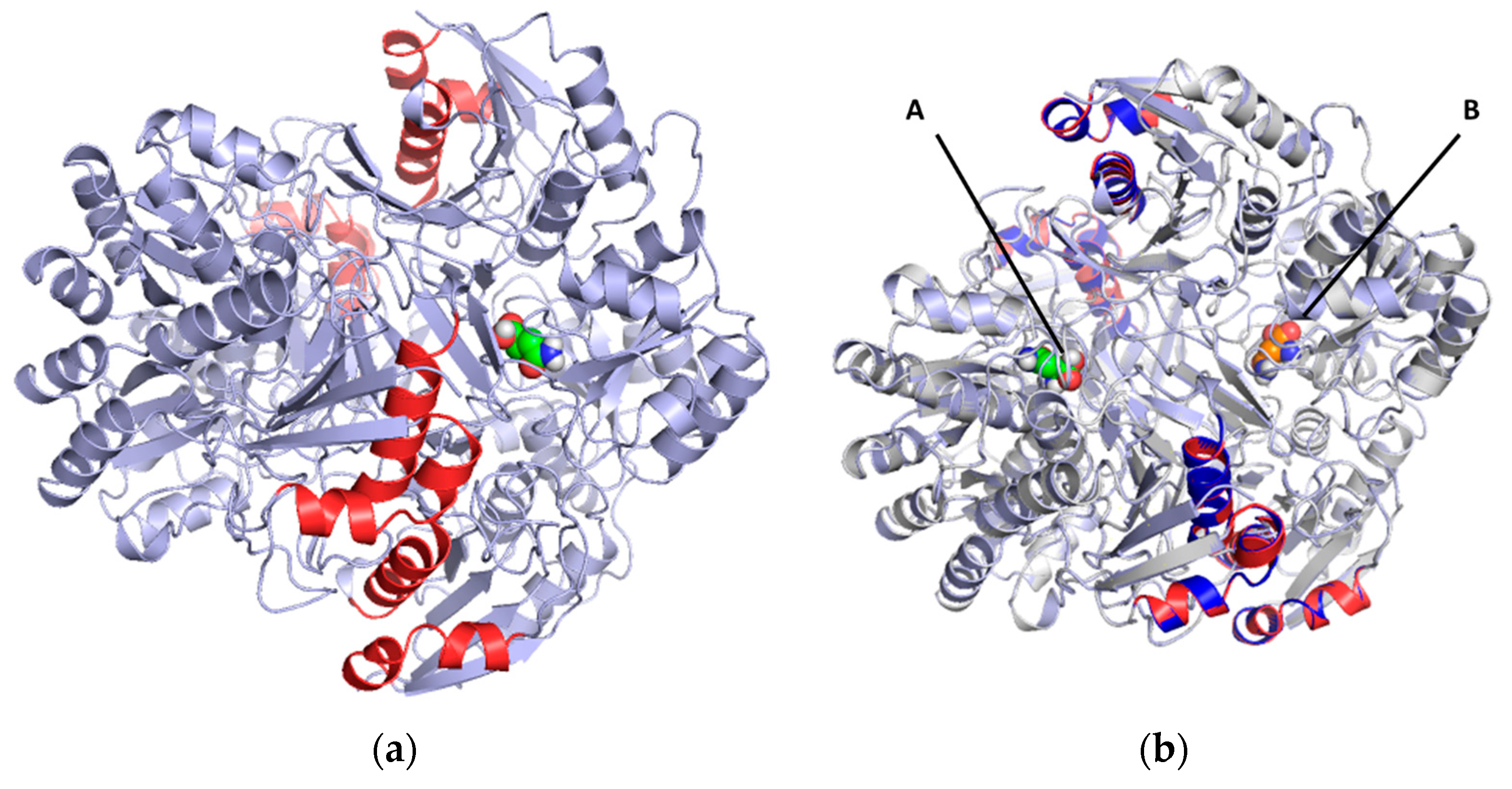
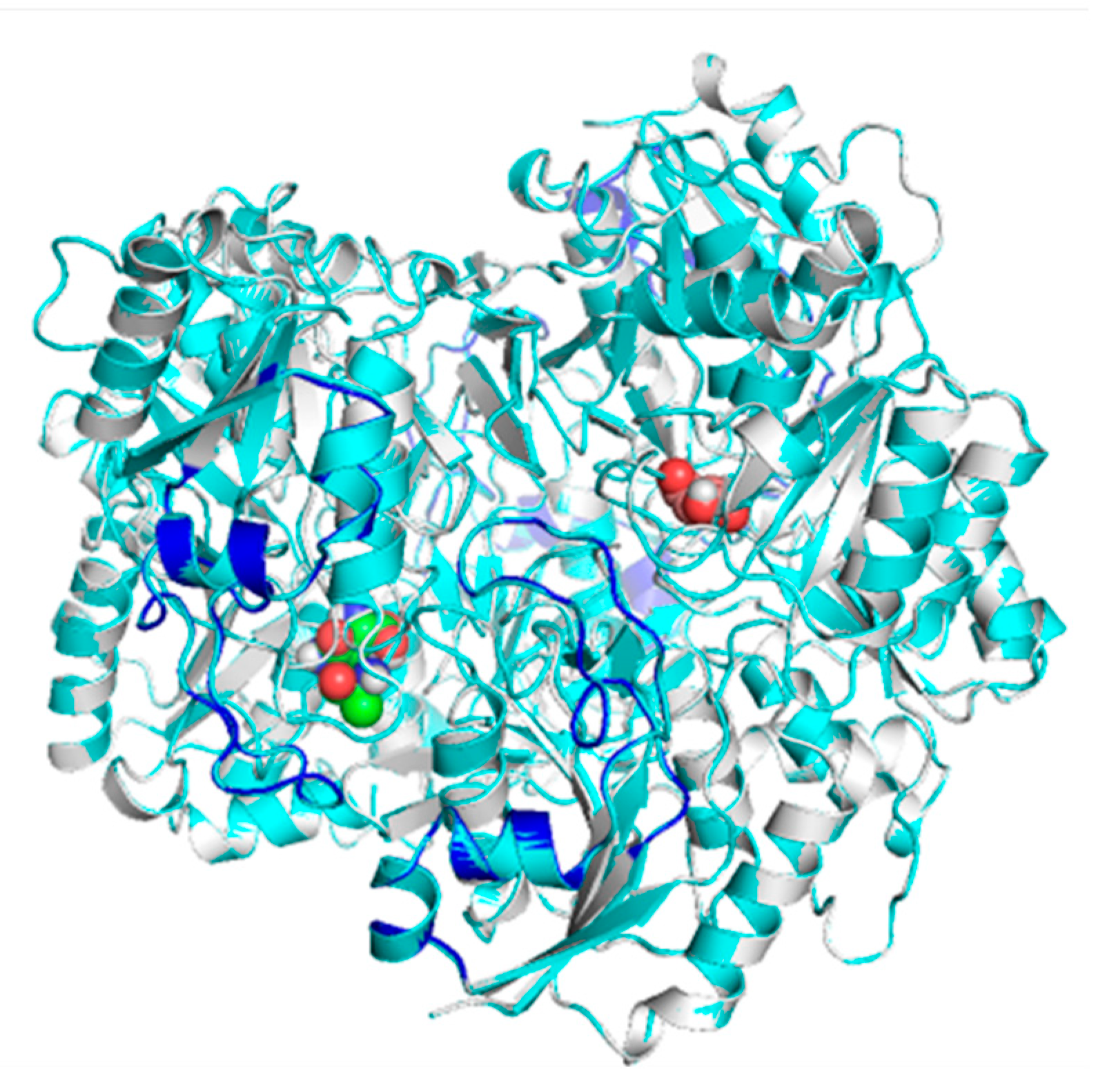
2.2. Mapping and Structure Determination of the Chimeric Enzyme
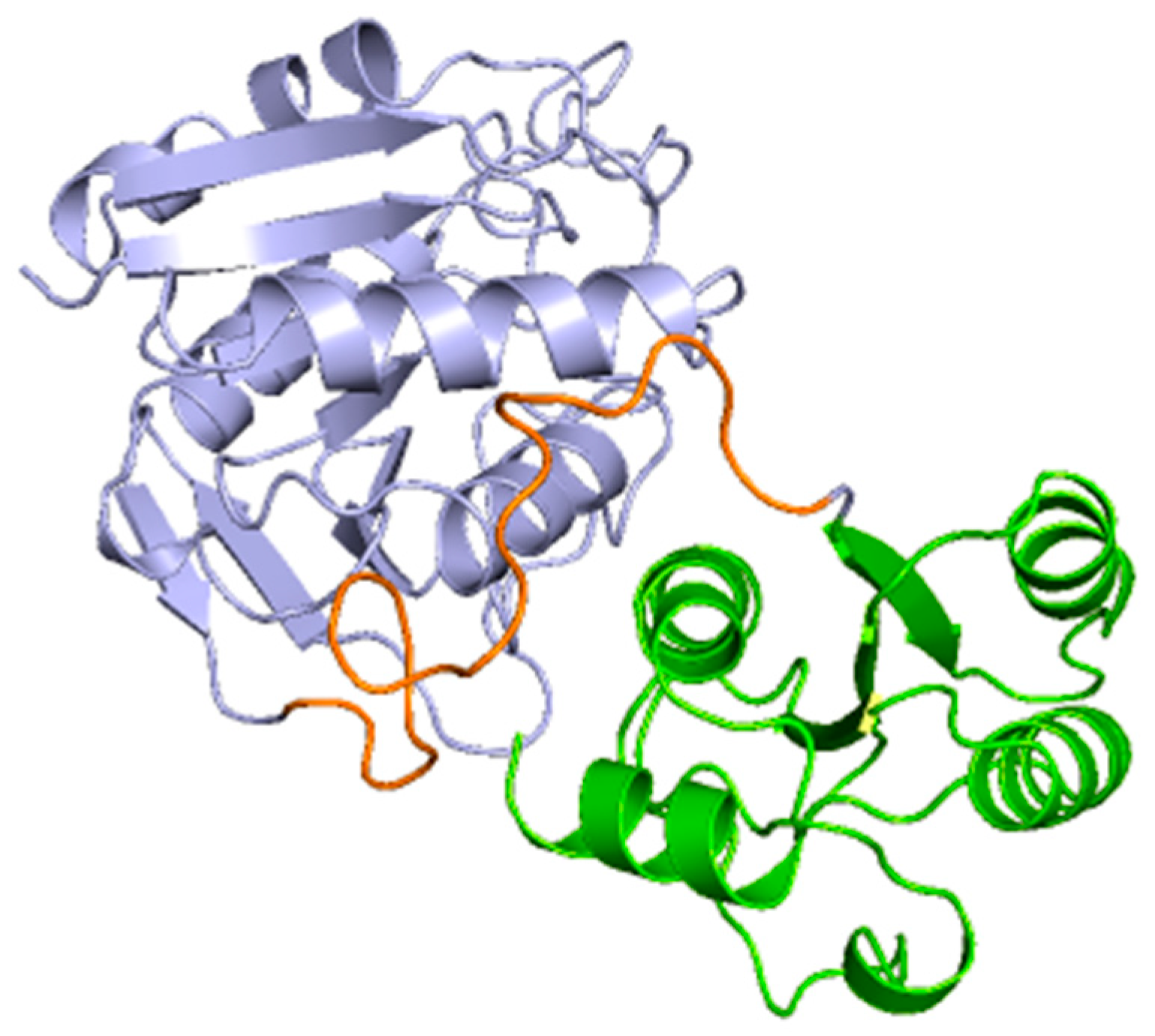
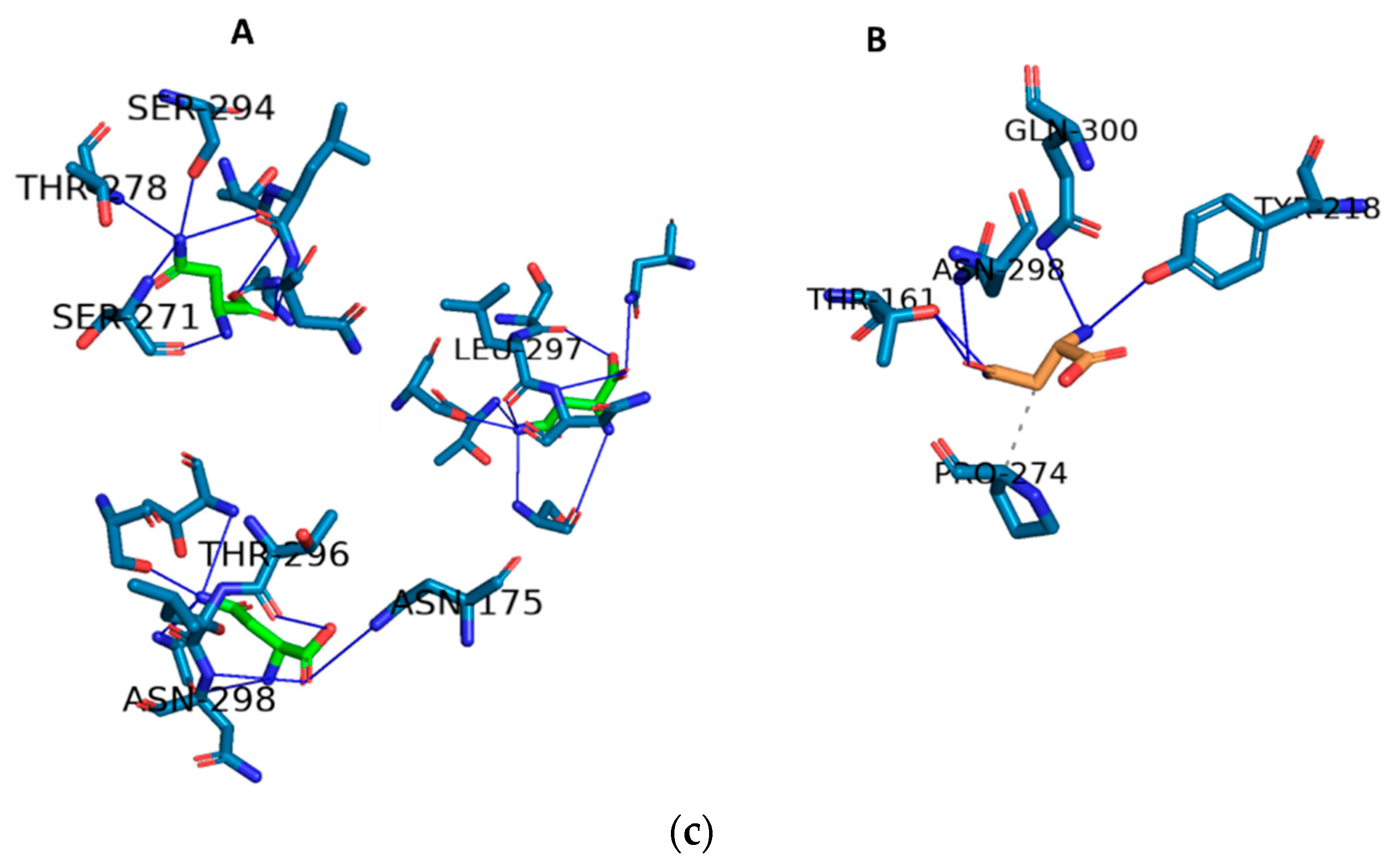
2.3. Docking and In Silico Determination of Substrate Affinity by the Chimeric Enzyme
3. Materials and Methods
3.1. Sequence Data of L-asparaginases
3.2. T-Cell Epitope Prediction and Epitope Density Determination
3.3. Prediction of Allergenic Epitopes
3.4. Epitope Alignment and Mapping
3.5. Modeling of the Three-Dimensional Structure of the Chimeric Enzyme
Verify3D Model
3.6. In Silico Determination of Substrate Affinity
3.7. Statistical Analysis of Epitope Density
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- de la Flor Brú Dra, J.T.; de la Calle, C.D.E.; Dra, M.I.H.V.; Pozo Román Vocales Regionales, J.; de la SEPEAP, J.D.; de Trabajo, G.; Bibliográficas, A.J. López Ávila Asma Alergia J Pellegrini Belinchón Docencia MIR Dra O González Calderón Educación para la Salud Promoción del Desarrollo Psicoemocional PJ Ruiz Lázaro. Consejo Editorial Subdirectores Ejecutivos Pediatría Integral. 2016. Available online: www.sepeap.org (accessed on 10 June 2022).
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Schmiegelow, K.; Forestier, E.; Hellebostad, M.; Heyman, M.; Kristinsson, J.; Söderhäll, S.; Taskinen, M. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010, 24, 345–354. [Google Scholar] [CrossRef]
- Batool, T.; Makky, E.A.; Jalal, M.; Yusoff, M.M. A Comprehensive Review on l-Asparaginase and Its Applications. Appl. Biochem. Biotechnol. 2016, 178, 900–923. [Google Scholar] [CrossRef] [PubMed]
- Müller, H. Use of L-asparaginase in childhood ALL. Crit. Rev. Oncol. 1998, 28, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, L.J.; Ettinger, A.G.; Avramis, V.I.; Gaynon, P.S. Acute Lymphoblastic Leukaemia. Biodrugs 1997, 7, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Batra, S.; Zhang, J. Asparagine: A Metabolite to Be Targeted in Cancers. Metabolites 2021, 11, 402. [Google Scholar] [CrossRef]
- Mehta, R.K.; Verma, S.; Pati, R.; Sengupta, M.; Khatua, B.; Jena, R.K.; Sethy, S.; Kar, S.K.; Mandal, C.; Roehm, K.H.; et al. Mutations in Subunit Interface and B-Cell Epitopes Improve Antileukemic Activities of Escherichia coli Asparaginase-II: Evaluation of Immunogenicity in Mice. J. Biol. Chem. 2014, 289, 3555–3570. [Google Scholar] [CrossRef]
- Pieters, R.; Hunger, S.P.; Boos, J.; Rizzari, C.; Silverman, L.; Baruchel, A.; Goekbuget, N.; Schrappe, M.; Pui, C.-H. L-asparaginase treatment in acute lymphoblastic leukemia. Cancer 2011, 117, 238–249. [Google Scholar] [CrossRef]
- Völler, S.; Pichlmeier, U.; Zens, A.; Hempel, G. Pharmacokinetics of recombinant asparaginase in children with acute lymphoblastic leukemia. Cancer Chemother. Pharmacol. 2018, 81, 305–314. [Google Scholar] [CrossRef]
- Ettinger, A.R. Pegaspargase (Oncaspar). J. Pediatr. Oncol. Nurs. 1995, 12, 46–48. [Google Scholar] [CrossRef]
- Kloos, R.; Van Der Sluis, I.M.; Mastrobattista, E.; Hennink, W.; Pieters, R.; Verhoef, J. Acute lymphoblastic leukaemia patients treated with PEGasparaginase develop antibodies to PEG and the succinate linker. Br. J. Haematol. 2019, 189, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Avramis, V.I.; Tiwari, P.N. Asparaginase pharmacokinetics and implications of therapeutic drug monitoring. In Leukemia and Lymphoma. Int. J. Nanomed. 2006, 1, 241–254. [Google Scholar]
- Lin, T.; Dumas, T.; Kaullen, J.; Berry, N.S.; Choi, M.R.; Zomorodi, K.; Silverman, J.A. Population Pharmacokinetic Model Development and Simulation for Recombinant Erwinia Asparaginase Produced in Pseudomonas fluorescens (JZP-458). Clin. Pharmacol. Drug Dev. 2021, 10, 1503–1513. [Google Scholar] [CrossRef]
- Krishna, M.; Nadler, S.G. Immunogenicity to Biotherapeutics—The Role of Anti-Drug Immune Complexes. Front. Immunol. 2016, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.H.G.; Fiúza, T.D.S.; Bath de Morais, S.; Souza, T.D.A.C.B.D.; Trevizani, R. Circumventing the side effects of L-asparaginase. Biomed. Pharmacother. 2021, 139, 111616. [Google Scholar] [CrossRef] [PubMed]
- Fung, M.K.L.; Chan, G.C.-F. Drug-induced amino acid deprivation as strategy for cancer therapy. J. Hematol. Oncol. 2017, 10, 144. [Google Scholar] [CrossRef]
- Avramis, V.I.; Tiwari, P.N. Asparaginase (native ASNase or pegylated ASNase) in the treatment of acute lymphoblastic leukemia. Int. J. Nanomed. 2006, 1, 241–254. Available online: https://pubmed.ncbi.nlm.nih.gov/17717965 (accessed on 12 June 2022).
- Offman, M.N.; Krol, M.; Patel, N.; Krishnan, S.; Liu, J.; Saha, V.; Bates, P. Lymphoid neoplasia rational engineering of L-asparaginase reveals importance of dual activity for cancer cell toxicity. Blood 2011, 117, 1614–1621. [Google Scholar] [CrossRef]
- Peterson, R.G.; Handschumacher, R.E.; Mitchell, M.S. Immunological responses to l-asparaginase. J. Clin. Investig. 1971, 50, 1080–1090. [Google Scholar] [CrossRef]
- Asselin, B.L.; Fisher, V. Impact of Clinical and Subclinical Hypersensitivity to Asparaginase in Acute Lymphoblastic Leukemia. Clin. J. Oncol. Nurs. 2014, 18, E107–E112. [Google Scholar] [CrossRef]
- Bowman, W.P.; Larsen, E.L.; Devidas, M.; Linda, S.B.; Blach, L.; Carroll, A.J.; Carroll, W.L.; Pullen, D.J.; Shuster, J.; Willman, C.L.; et al. Augmented therapy improves outcome for pediatric high risk acute lymphocytic leukemia: Results of Children’s Oncology Group trial P9906. Pediatr. Blood Cancer 2011, 57, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.A.; Smith, C.A.; Yang, W.; Daté, M.; Bashford, D.; Larsen, E.C.; Bowman, W.P.; Liu, C.; Ramsey, L.; Chang, T.; et al. HLA-DRB1*07:01 is associated with a higher risk of asparaginase allergies. Blood 2014, 124, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Belviso, S.; Iuliano, R.; Amato, R.; Perrotti, N.; Menniti, M. The human asparaginase enzyme (ASPG) inhibits growth in leukemic cells. PLoS ONE 2017, 12, e0178174. [Google Scholar] [CrossRef]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Schalk, A.M.; Nguyen, H.-A.; Rigouin, C.; Lavie, A. Identification and Structural Analysis of an l-Asparaginase Enzyme from Guinea Pig with Putative Tumor Cell Killing Properties. J. Biol. Chem. 2014, 289, 33175–33186. [Google Scholar] [CrossRef] [PubMed]
- Pokrovsky, V.S.; Kazanov, M.D.; Dyakov, I.N.; Pokrovskaya, M.V.; Aleksandrova, S.S. Comparative immunogenicity and structural analysis of epitopes of different bacterial L-asparaginases. BMC Cancer 2016, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.L.; Perissinotti, A.J.; Bixby, D.L.; Brown, J.; Burke, P.W. Catalyzing improvements in ALL therapy with asparaginase. Blood Rev. 2017, 31, 328–338. [Google Scholar] [CrossRef]
- Vrooman, L.M.; Supko, J.G.; Neuberg, D.S.; Asselin, B.L.; Athale, U.H.; Clavell, L.; Kelly, K.M.; Laverdière, C.; Michon, B.; Schorin, M.; et al. Erwinia asparaginase after allergy to E. coli asparaginase in children with acute lymphoblastic leukemia. Pediatr. Blood Cancer 2010, 54, 199–205. [Google Scholar] [CrossRef]
- Zha, D. Glycoengineered Pichia-Based Expression of Monoclonal Antibodies. In Glycosylation Engineering of Biopharmaceuticals; Springer: Berlin/Heidelberg, Germany, 2013; pp. 31–43. [Google Scholar] [CrossRef]
- Effer, B.; Lima, G.M.; Cabarca, S.; Pessoa, A.; Farías, J.G.; Monteiro, G. L-Asparaginase from E. chrysanthemi expressed in glycoswitch®: Effect of His-Tag fusion on the extracellular expression. Prep. Biochem. Biotechnol. 2019, 49, 679–685. [Google Scholar] [CrossRef]
- Moola, Z.B.; Scawen, M.D.; Atkinson, T.; Nicholls, D.J. Erwinia chrysanthemi l-asparaginase: Epitope mapping and production of antigenically modified enzymes. Biochem. J. 1994, 302, 921–927. [Google Scholar] [CrossRef]
- Thomas, X.; Le Jeune, C. Erythrocyte encapsulated l-asparaginase (GRASPA) in acute leukemia. Int. J. Hematol. Oncol. 2016, 5, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J.; Zalewska-Szewczyk, B. Hypersensitivity reactions to asparaginase therapy in acute lymphoblastic leukemia: Immunology and clinical consequences. Futur. Oncol. 2022, 18, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Newsted, W.; Ramjeesingh, M.; Zywulko, M.; Rothstein, S.; Shami, E. Engineering resistance to trypsin inactivation into l-asparaginase through the production of a chimeric protein between the enzyme and a protective single-chain antibody. Enzym. Microb. Technol. 1995, 17, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Gaofu, Q.; Rongyue, C.; Dan, M.; Xiuyun, Z.; Xuejun, W.; Jie, W.; Jingjing, L. Asparaginase Display of Human Cholesteryl Ester Transfer Protein (CETP) B Cell Epitopes for Inducing High Titers of Anti-CETP Antibodies in Vivo. Protein Pept. Lett. 2006, 13, 149–154. [Google Scholar] [CrossRef]
- Belén, L.H.; Lissabet, J.B.; Rangel-Yagui, C.D.O.; Effer, B.; Monteiro, G.; Pessoa, A.; Avendaño, J.G.F. A structural in silico analysis of the immunogenicity of l-asparaginase from Escherichia coli and Erwinia carotovora. Biologicals 2019, 59, 47–55. [Google Scholar] [CrossRef]
- Belén, L.H.; Lissabet, J.F.B.; Rangel-Yagui, C.D.O.; Monteiro, G.; Pessoa, A.; Farías, J.G. Immunogenicity assessment of fungal l-asparaginases: An in silico approach. SN Appl. Sci. 2020, 2, 222. [Google Scholar] [CrossRef]
- Pokrovskaya, M.V.; Pokrovsky, V.S.; Aleksandrova, S.S.; Sokolov, N.N.; Zhdanov, D.D. Molecular Analysis of L-Asparaginases for Clarification of the Mechanism of Action and Optimization of Pharmacological Functions. Pharmaceutics 2022, 14, 599. [Google Scholar] [CrossRef]
- Beckett, A.; Gervais, D. What makes a good new therapeutic l-asparaginase? World J. Microbiol. Biotechnol. 2019, 35, 152. [Google Scholar] [CrossRef]
- Rigouin, C.; Nguyen, H.A.; Schalk, A.M.; Lavie, A. Discovery of human-like L-asparaginases with potential clinical use by directed evolution. Sci. Rep. 2017, 7, 10224. [Google Scholar] [CrossRef]
- Lissabet, J.F.B. A large-scale immunoinformatics analysis of the human papillomaviruses reveals a common E5 oncoprotein-pattern to evade the immune response. Gene Rep. 2018, 10, 1–6. [Google Scholar] [CrossRef]
- Feldmann, M.; Howard, J.G.; Desaymard, C. Role of Antigen Structure in the Discrimination between Tolerance and Immunity by B Cells. Immunol. Rev. 1975, 23, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, Y.-H. High epitope density in a single protein molecule significantly enhances antigenicity as well as immunogenicity: A novel strategy for modern vaccine development and a preliminary investigation about B cell discrimination of monomeric proteins. Eur. J. Immunol. 2005, 35, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Kellogg, C.; Gutiérrez, A.H.; Moise, L.; Terry, F.; Martin, W.D.; De Groot, A.S. CHOPPI: A web tool for the analysis of immunogenicity risk from host cell proteins in CHO-based protein production. Biotechnol. Bioeng. 2014, 111, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Moise, L.; Liu, R.; Gutierrez, A.H.; Tassone, R.; Bailey-Kellogg, C.; Martin, W. Immune camouflage: Relevance to vaccines and human immunology. Hum. Vaccines Immunother. 2014, 10, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- He, L.; De Groot, A.S.; Gutierrez, A.H.; Martin, W.D.; Moise, L.; Bailey-Kellogg, C. Integrated assessment of predicted MHC binding and cross-conservation with self reveals patterns of viral camouflage. BMC Bioinform. 2014, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Narta, U.K.; Kanwar, S.S.; Azmi, W. Pharmacological and clinical evaluation of l-asparaginase in the treatment of leukemia. Crit. Rev. Oncol. 2007, 61, 208–221. [Google Scholar] [CrossRef]
- D’iakov, I.N.; Pokrovskiĭ, V.S.; Sannikova, E.P.; Bulushova, N.V.; Pokrovskaia, M.V.; Aleksandrova, S.S. Cross-immunogenicity of various bacterial L-asparaginases. Zh Mikrobiol. Epidemiol. Immunobiol. 2014, 6, 100–104. Available online: http://europepmc.org/abstract/MED/25816523 (accessed on 10 June 2022).
- Sharma, D.; Singh, K.; Singh, K.; Mishra, A. Insights into the Microbial L-Asparaginases: From Production to Practical Applications. Curr. Protein. Pept. Sci. 2019, 20, 452–464. [Google Scholar] [CrossRef]
- Lissabet, J.F.B.; Belén, L.H.; Lee-Estevez, M.; Risopatrón, J.; Valdebenito, I.; Figueroa, E.; Farias, J. The CatSper channel is present and plays a key role in sperm motility of the Atlantic salmon (Salmo salar). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2020, 241, 110634. [Google Scholar] [CrossRef]
- Andreatta, M.; Karosiene, E.; Rasmussen, M.; Stryhn, A.; Buus, S.; Nielsen, M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015, 67, 641–650. [Google Scholar] [CrossRef]
- Evans, W.E.; Tsiatis, A.; Rivera, G.; Murphy, S.B.; Dahl, G.V.; Denison, M.; Crom, W.R.; Barker, L.F.; Mauer, A.M. Anaphylactoid reactions to Escherichia coli and Erwinia asparaginase in children with leukemia and lymphoma. Cancer 1982, 49, 1378–1383. [Google Scholar] [CrossRef]
- Körholz, D.; Urbanek, R.; Nürnberger, W.; Jobke, A.; Göbel, U.; Wahn, V. Formation of specific IgG antibodies in l-asparaginase treatment. Distribution of IgG subclasses. Mon. Kinderheilkd 1987, 135, 325–328. Available online: http://europepmc.org/abstract/MED/3475571 (accessed on 10 August 2022).
- Swain, A.L.; Jaskólski, M.; Housset, D.; Rao, J.K.; Wlodawer, A. Crystal structure of Escherichia coli L-asparaginase, an enzyme used in cancer therapy. Proc. Natl. Acad. Sci. USA 1993, 90, 1474–1478. [Google Scholar] [CrossRef]
- Pelton, J.T.; McLean, L.R. Spectroscopic Methods for Analysis of Protein Secondary Structure. Anal. Biochem. 2000, 277, 167–176. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, T.S.; Scapin, S.M.; de Andrade, W.; Fasciotti, M.; de Magalhães, M.T.; Almeida, M.S.; Lima, L.M.T. Biophysical characterization of two commercially available preparations of the drug containing Escherichia coli L-Asparaginase 2. Biophys. Chem. 2021, 271, 106554. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K. The prospects and opportunities of protein structure prediction with AI. Nat. Rev. Mol. Cell Biol. 2022, 23, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35 (Suppl. S2), W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Jakovlic, I.; Wang, W.-M. In silico characterisation, homology modelling and structure-based functional annotation of blunt snout bream (Megalobrama amblycephala) Hsp70 and Hsc70 proteins. J. Anim. Sci. Technol. 2015, 57, 44. [Google Scholar] [CrossRef]
- Singh, R.; Gurao, A.; Rajesh, C.; Mishra, S.K.; Rani, S.; Behl, A.; Kumar, V.; Kataria, R.S. Comparative modeling and mutual docking of structurally uncharacterized heat shock protein 70 and heat shock factor-1 proteins in water buffalo. Vet. World 2019, 12, 2036. [Google Scholar] [CrossRef]
- Nomme, J.; Su, Y.; Lavie, A. Elucidation of the Specific Function of the Conserved Threonine Triad Responsible for Human l-Asparaginase Autocleavage and Substrate Hydrolysis. J. Mol. Biol. 2014, 426, 2471–2485. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; Christmas, S.; Middleton, D.; Jones, A.R. Allele frequency net: A database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011, 39, D913–D919. [Google Scholar] [CrossRef] [PubMed]
- Wiesch, J.S.Z.; Lauer, G.M.; Day, C.L.; Kim, A.Y.; Ouchi, K.; Duncan, J.E.; Wurcel, A.G.; Timm, J.; Jones, A.M.; Mothe, B.; et al. Broad Repertoire of the CD4+ Th Cell Response in Spontaneously Controlled Hepatitis C virus Infection Includes Dominant and Highly Promiscuous Epitopes. J. Immunol. 2005, 175, 3603–3613. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega. Curr. Protoc. Bioinform. 2014, 48, 3.13.1–3.13.16. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Dym, O.; Eisenberg, D.; Yeates, T.O. Detection of Errors in Protein Models. In International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules; Springer: Dordrecht, The Netherland, 2006. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Bhattarai, A.; Wang, J.; Miao, Y. Retrospective ensemble docking of allosteric modulators in an adenosine G-protein-coupled receptor. Biochim. Et Biophys. Acta BBA Gen. Subj. 2020, 1864, 129615. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedroso, A.; Herrera Belén, L.; Beltrán, J.F.; Castillo, R.L.; Pessoa, A.; Pedroso, E.; Farías, J.G. In Silico Design of a Chimeric Humanized L-asparaginase. Int. J. Mol. Sci. 2023, 24, 7550. https://doi.org/10.3390/ijms24087550
Pedroso A, Herrera Belén L, Beltrán JF, Castillo RL, Pessoa A, Pedroso E, Farías JG. In Silico Design of a Chimeric Humanized L-asparaginase. International Journal of Molecular Sciences. 2023; 24(8):7550. https://doi.org/10.3390/ijms24087550
Chicago/Turabian StylePedroso, Alejandro, Lisandra Herrera Belén, Jorge F. Beltrán, Rodrigo L. Castillo, Adalberto Pessoa, Enrique Pedroso, and Jorge G. Farías. 2023. "In Silico Design of a Chimeric Humanized L-asparaginase" International Journal of Molecular Sciences 24, no. 8: 7550. https://doi.org/10.3390/ijms24087550