

Interleukin 26 Induces Macrophage IL-9 Expression in Rheumatoid Arthritis
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. IL-26+ and IL-9+ Macrophages Were Highly Expressed in Synovial Tissue in RA
2.2. Effect of IL-26 on IL-9 Cytokines Expression and CD80 Macrophage Differentiation in RAW264.7
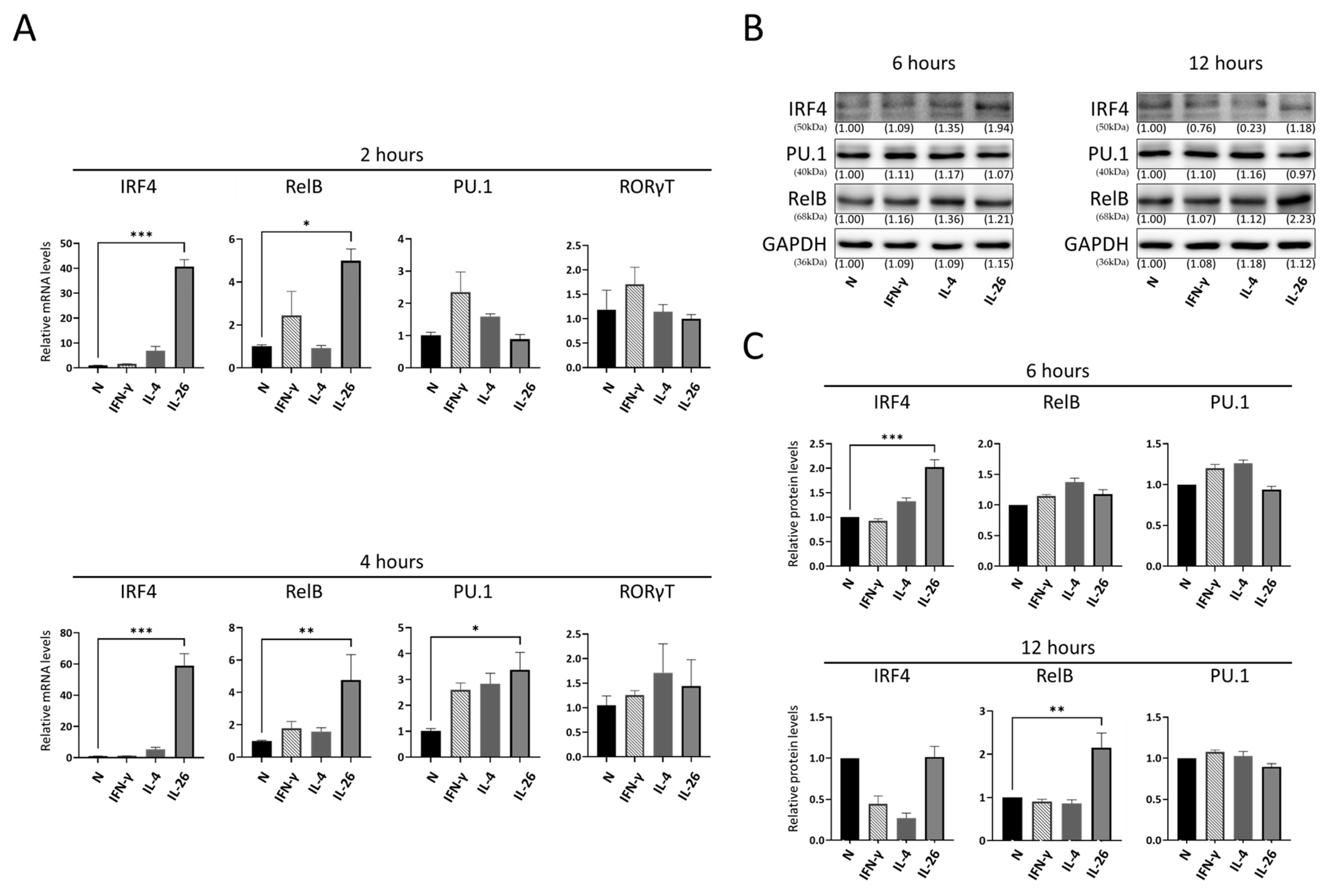
2.3. Effect of IL-26 on IL-9, IL-17A-Related Transcription Factor Expression in RAW264.7
2.4. Effect of IL-26 on AKT and FoxO1 Activation in RAW264.7 and THP-1
2.5. Effect of IL-26 plus MK2206 on IL-9- and IL17A-Regulated Transcriptional Gene Expression in RAW264.7 and THP-1
2.6. Effects of IL-26 plus MK2206 on IL-9 and IL-17A Cytokines Expression in RAW264.7, THP-1, and Primary Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Hematoxylin and Eosin (H&E) and Immunofluorescence (IF) Staining
4.3. Cell Lines and Primary Culture
4.4. Flow Cytometry
4.5. Protein Extraction
4.6. Western Blot
4.7. RNA Extraction and Real-Time PCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scott, D.L.; Pugner, K.; Kaarela, K.; Doyle, D.V.; Woolf, A.; Holmes, J.; Hieke, K. The links between joint damage and disability in rheumatoid arthritis. Rheumatol. Oxf. Engl. 2000, 39, 122–132. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, Z.; Chang, C.; Lu, L.; Lau, C.S.; Lu, Q. Th9 cells and IL-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum. Immunol. 2017, 78, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Takayanagi, H. Inflammation and bone destruction in arthritis: Synergistic activity of immune and mesenchymal cells in joints. Front. Immunol. 2012, 3, 77. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Ferrante, A.; Cipriani, P.; Giacomelli, R.; Triolo, G. Interleukin-9 and T helper type 9 cells in rheumatic diseases. Clin. Exp. Immunol. 2016, 185, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tanida, S.; Yoshitomi, H.; Nishitani, K.; Ishikawa, M.; Kitaori, T.; Ito, H.; Nakamura, T. CCL20 produced in the cytokine network of rheumatoid arthritis recruits CCR6+ mononuclear cells and enhances the production of IL-6. Cytokine 2009, 47, 112–118. [Google Scholar] [CrossRef]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Fossiez, F.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Chabaud, M.; Fossiez, F.; Taupin, J.L.; Miossec, P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 1998, 161, 409–414. [Google Scholar] [CrossRef]
- Corvaisier, M.; Delneste, Y.; Jeanvoine, H.; Preisser, L.; Blanchard, S.; Garo, E.; Hoppe, E.; Barre, B.; Audran, M.; Bouvard, B.; et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. 2012, 10, e1001395. [Google Scholar] [CrossRef]
- Stephen-Victor, E.; Fickenscher, H.; Bayry, J. IL-26: An Emerging Proinflammatory Member of the IL-10 Cytokine Family with Multifaceted Actions in Antiviral, Antimicrobial, and Autoimmune Responses. PLoS Pathog. 2016, 12, e1005624. [Google Scholar] [CrossRef] [PubMed]
- Louhaichi, S.; Mlika, M.; Hamdi, B.; Hamzaoui, K.; Hamzaoui, A. Sputum IL-26 Is Overexpressed in Severe Asthma and Induces Proinflammatory Cytokine Production and Th17 Cell Generation: A Case-Control Study of Women. J. Asthma. Allergy 2020, 13, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Lai, X.; Li, J.; Huang, L.; Liu, Y.; Cao, J. Interleukin-26 is overexpressed in human sepsis and contributes to inflammation, organ injury, and mortality in murine sepsis. Crit. Care Lond. Engl. 2019, 23, 290. [Google Scholar] [CrossRef] [PubMed]
- Bartziokas, K.; Fouka, E.; Loukides, S.; Steiropoulos, P.; Bakakos, P.; Papaioannou, A.I. IL-26 in the Lung and Its Role in COPD Inflammation. J. Pers. Med. 2022, 12, 1685. [Google Scholar] [CrossRef]
- Cardenas, E.I.; Ekstedt, S.; Piersiala, K.; Petro, M.; Karlsson, A.; Kågedal, Å.; Kumlien Georén, S.; Cardell, L.O.; Lindén, A. Increased IL-26 associates with markers of hyperinflammation and tissue damage in patients with acute COVID-19. Front. Immunol. 2022, 13, 1016991. [Google Scholar] [CrossRef]
- Koch, S.; Sopel, N.; Finotto, S. Th9 and other IL-9-producing cells in allergic asthma. Semin. Immunopathol. 2017, 39, 55–68. [Google Scholar] [CrossRef]
- Knappe, A.; Hör, S.; Wittmann, S.; Fickenscher, H. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J. Virol. 2000, 74, 3881–3887. [Google Scholar] [CrossRef]
- Hör, S.; Pirzer, H.; Dumoutier, L.; Bauer, F.; Wittmann, S.; Sticht, H.; Renauld, J.C.; de Waal Malefyt, R.; Fickenscher, H. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J. Biol. Chem. 2004, 279, 33343–33351. [Google Scholar] [CrossRef]
- Sheikh, F.; Baurin, V.V.; Lewis-Antes, A.; Shah, N.K.; Smirnov, S.V.; Anantha, S.; Dickensheets, H.; Dumoutier, L.; Renauld, J.C.; Zdanov, A.; et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J. Immunol. 2004, 172, 2006–2010. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Sheikh, F.; Dickensheets, H.; Savan, R.; Young, H.A.; Walter, M.R. Interleukin-26: An IL-10-related cytokine produced by Th17 cells. Cytokine. Growth Factor Rev. 2010, 21, 393–401. [Google Scholar] [CrossRef]
- Itoh, T.; Hatano, R.; Komiya, E.; Otsuka, H.; Narita, Y.; Aune, T.M.; Dang, N.H.; Matsuoka, S.; Naito, H.; Tominaga, M.; et al. Biological Effects of IL-26 on T Cell–Mediated Skin Inflammation, Including Psoriasis. J. Investig. Dermatol. 2019, 139, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.J.; Wang, C.Y.; Lin, Y.H.; Lin, G.J.; Huang, S.H.; Shyu, J.F.; Sytwu, H.K.; Cheng, C.P. Interleukin 26 suppresses receptor activator of nuclear factor kappaB ligand induced osteoclastogenesis via down-regulation of nuclear factor of activated T-cells, cytoplasmic 1 and nuclear factor kappaB activity. Rheumatol. Oxf. Engl. 2016, 55, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Wang, Y.H.; Peng, Y.J.; Liu, F.C.; Lin, G.J.; Huang, S.H.; Sytwu, H.K.; Cheng, C.P. Interleukin 26 Skews Macrophage Polarization Towards M1 Phenotype by Activating cJUN and the NF-κB Pathway. Cells 2020, 9, 938. [Google Scholar] [CrossRef] [PubMed]
- Grailer, J.; Bosmann, M.; Ward, P. Regulatory effects of C5a on IL-17A, IL-17F, and IL-23. Front. Immunol. 2013, 3, 387. [Google Scholar] [CrossRef]
- Tong, H.; Feng, H.; Hu, X.; Wang, M.-F.; Song, Y.-F.; Wen, X.-L.; Li, Y.-R.; Wan, X.-P. Identification of Interleukin-9 Producing Immune Cells in Endometrial Carcinoma and Establishment of a Prognostic Nomogram. Front. Immunol. 2020, 11, 544248. [Google Scholar] [CrossRef]
- Bosmann, M.; Sarma, J.V.; Atefi, G.; Zetoune, F.S.; Ward, P.A. Evidence for anti-inflammatory effects of C5a on the innate IL-17A/IL-23 axis. FASEB J. 2012, 26, 1640–1651. [Google Scholar] [CrossRef]
- Goswami, R.; Kaplan, M.H. A brief history of IL-9. J. Immunol. 2011, 186, 3283–3288. [Google Scholar] [CrossRef]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef]
- Malik, S.; Sadhu, S.; Elesela, S.; Pandey, R.P.; Chawla, A.S.; Sharma, D.; Panda, L.; Rathore, D.; Ghosh, B.; Ahuja, V.; et al. Transcription factor Foxo1 is essential for IL-9 induction in T helper cells. Nat. Commun. 2017, 8, 815. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Nakae, S.; Nambu, A.; Sudo, K.; Iwakura, Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 2003, 171, 6173–6177. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Manzo, A.; Vitolo, B.; La Manna, M.P.; Giardina, G.; Sireci, G.; Dieli, F.; Montecucco, C.M.; et al. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatol. Oxf. 2015, 54, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.; Kumar, U.; Das, S.; Chaudhuri, J.; Kumar, P.; Kanjilal, M.; Ghosh, P.; Sircar, G.; Basyal, R.K.; Kanga, U.; et al. Synovial IL-9 facilitates neutrophil survival, function and differentiation of Th17 cells in rheumatoid arthritis. Arthritis Res. 2018, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Richmond, A. Th9 and Th17 cells: The controversial twins in cancer immunity. J. Clin. Investig. 2020, 130, 3409–3411. [Google Scholar] [CrossRef]
- Salazar, Y.; Zheng, X.; Brunn, D.; Raifer, H.; Picard, F.; Zhang, Y.; Winter, H.; Guenther, S.; Weigert, A.; Weigmann, B.; et al. Microenvironmental Th9 and Th17 lymphocytes induce metastatic spreading in lung cancer. J. Clin. Investig. 2020, 130, 3560–3575. [Google Scholar] [CrossRef]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef]
- Tamiya, T.; Ichiyama, K.; Kotani, H.; Fukaya, T.; Sekiya, T.; Shichita, T.; Honma, K.; Yui, K.; Matsuyama, T.; Nakao, T.; et al. Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J. Immunol. 2013, 191, 2360–2371. [Google Scholar] [CrossRef]
- Hueber, A.J.; Asquith, D.L.; Miller, A.M.; Reilly, J.; Kerr, S.; Leipe, J.; Melendez, A.J.; McInnes, I.B. Cutting Edge: Mast Cells Express IL-17A in Rheumatoid Arthritis Synovium. J. Immunol. 2010, 184, 3336–3340. [Google Scholar] [CrossRef]
- Mudter, J.; Yu, J.; Zufferey, C.; Brustle, A.; Wirtz, S.; Weigmann, B.; Hoffman, A.; Schenk, M.; Galle, P.R.; Lehr, H.A.; et al. IRF4 regulates IL-17A promoter activity and controls RORgammat-dependent Th17 colitis in vivo. Inflamm. Bowel Dis. 2011, 17, 1343–1358. [Google Scholar] [CrossRef]
- Powolny-Budnicka, I.; Riemann, M.; Tanzer, S.; Schmid, R.M.; Hehlgans, T.; Weih, F. RelA and RelB transcription factors in distinct thymocyte populations control lymphotoxin-dependent interleukin-17 production in gammadelta T cells. Immunity 2011, 34, 364–374. [Google Scholar] [CrossRef]
- Malik, S.; Awasthi, A. Transcriptional Control of Th9 Cells: Role of Foxo1 in Interleukin-9 Induction. Front. Immunol. 2018, 9, 995. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.; Martin, B.; Luka, M.; Mir, L.; Auffray, C.; Lucas, B.; Bismuth, G.; Charvet, C. Foxo1 Is a T Cell-Intrinsic Inhibitor of the RORgammat-Th17 Program. J. Immunol. 2015, 195, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Zhao, Z.B.; Liu, Q.Z.; Hu, T.D.; Long, J.; Yan, K.; Lian, Z.X. FoxO1 is a regulator of MHC-II expression and anti-tumor effect of tumor-associated macrophages. Oncogene 2018, 37, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Ranjan, R.; Lee, Y.G.; Park, G.Y.; Karpurapu, M.; Deng, J.; Xiao, L.; Kim, J.Y.; Unterman, T.G.; Christman, J.W. Distinct role of FoxO1 in M-CSF- and GM-CSF-differentiated macrophages contributes LPS-mediated IL-10: Implication in hyperglycemia. J. Leukoc. Biol. 2015, 97, 327–339. [Google Scholar] [CrossRef]
- Fan, W.; Morinaga, H.; Kim, J.J.; Bae, E.; Spann, N.J.; Heinz, S.; Glass, C.K.; Olefsky, J.M. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 2010, 29, 4223–4236. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-H.; Peng, Y.-J.; Liu, F.-C.; Lin, G.-J.; Huang, S.-H.; Sytwu, H.-K.; Cheng, C.-P. Interleukin 26 Induces Macrophage IL-9 Expression in Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 7526. https://doi.org/10.3390/ijms24087526
Wang Y-H, Peng Y-J, Liu F-C, Lin G-J, Huang S-H, Sytwu H-K, Cheng C-P. Interleukin 26 Induces Macrophage IL-9 Expression in Rheumatoid Arthritis. International Journal of Molecular Sciences. 2023; 24(8):7526. https://doi.org/10.3390/ijms24087526
Chicago/Turabian StyleWang, Yi-Hsun, Yi-Jen Peng, Feng-Cheng Liu, Gu-Jiun Lin, Shing-Hwa Huang, Huey-Kang Sytwu, and Chia-Pi Cheng. 2023. "Interleukin 26 Induces Macrophage IL-9 Expression in Rheumatoid Arthritis" International Journal of Molecular Sciences 24, no. 8: 7526. https://doi.org/10.3390/ijms24087526