
Unlocking STING as a Therapeutic Antiviral Strategy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
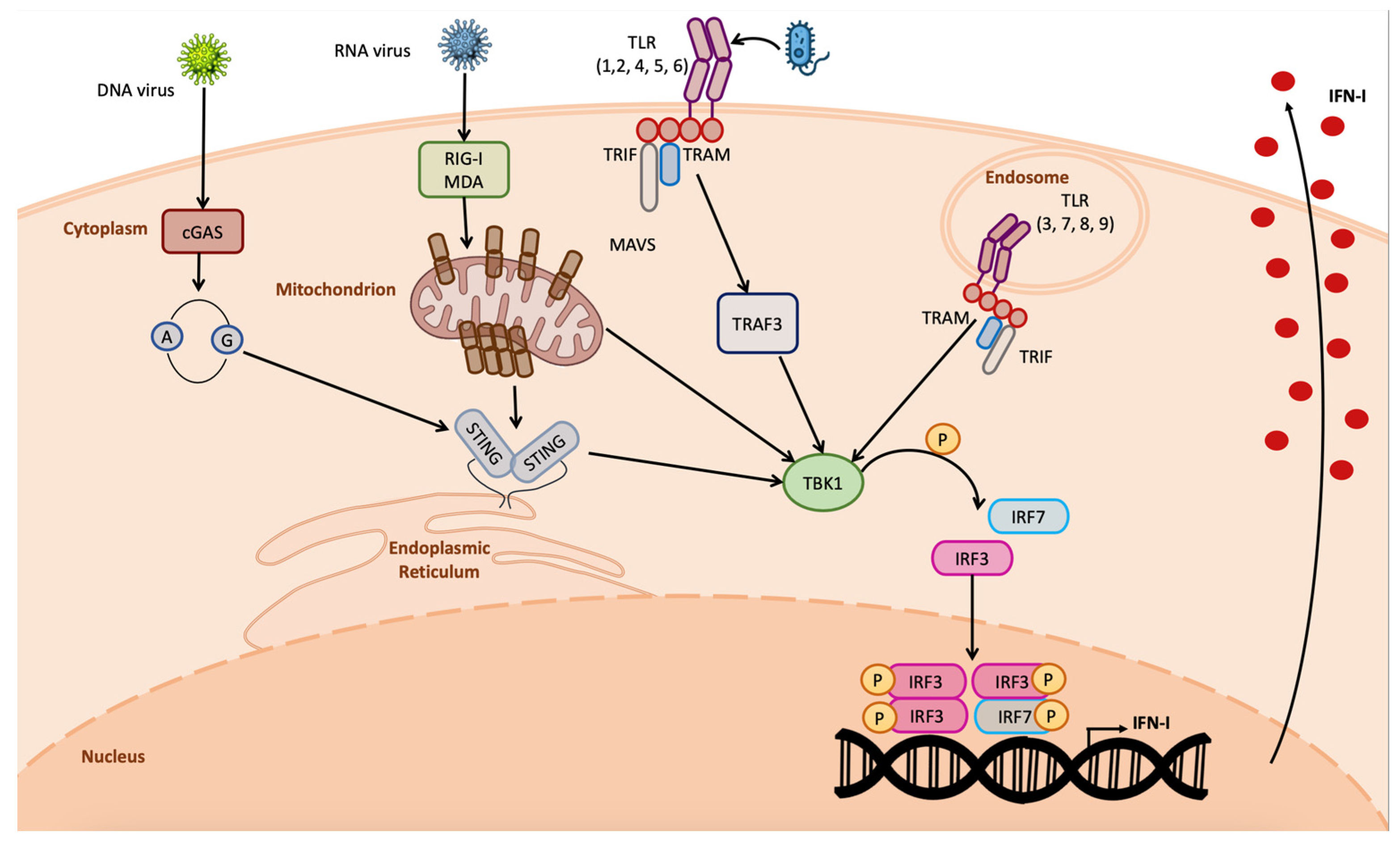
:1. Introduction
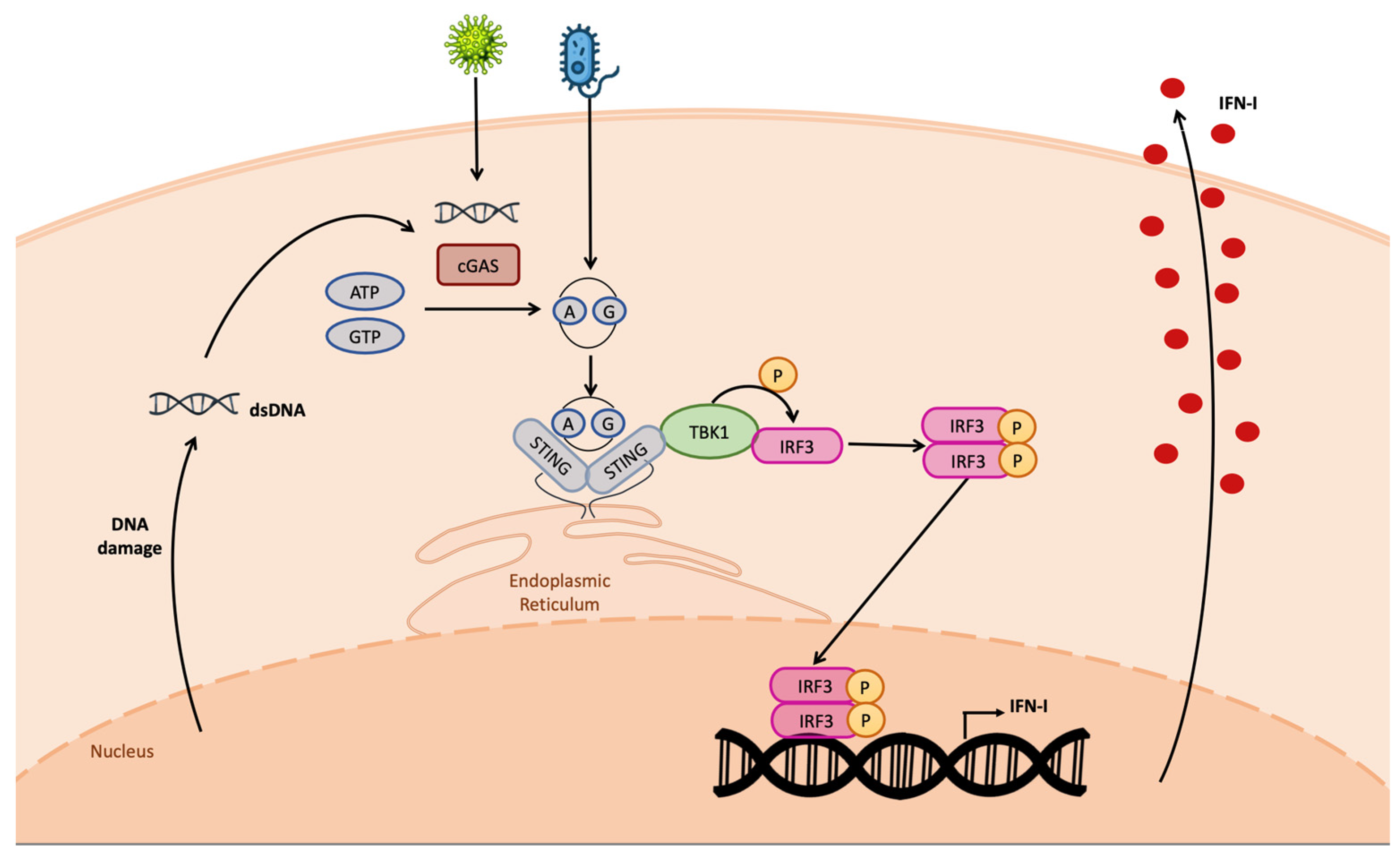
2. Functional Role of STING
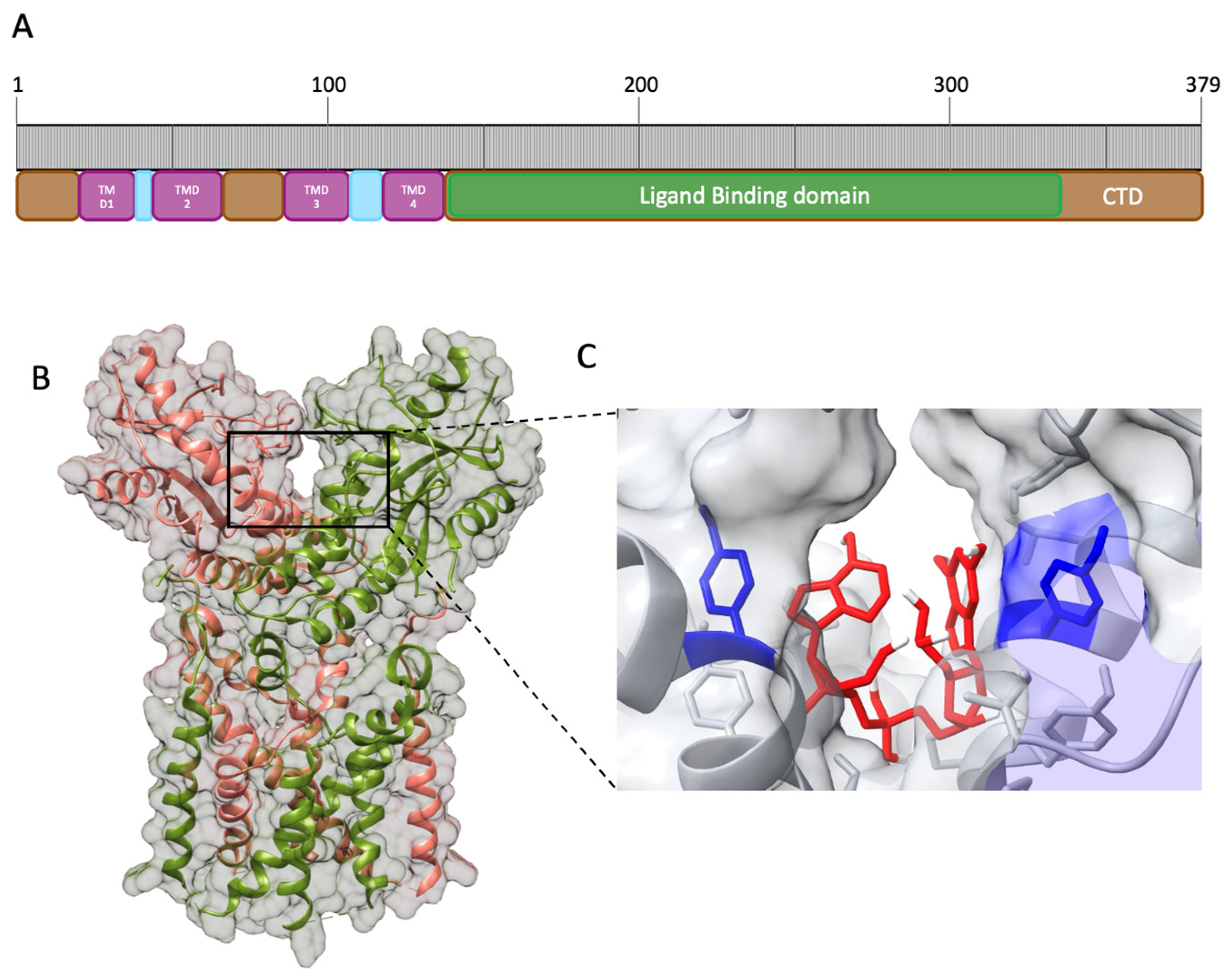
3. Structure of STING and Interaction with Ligands
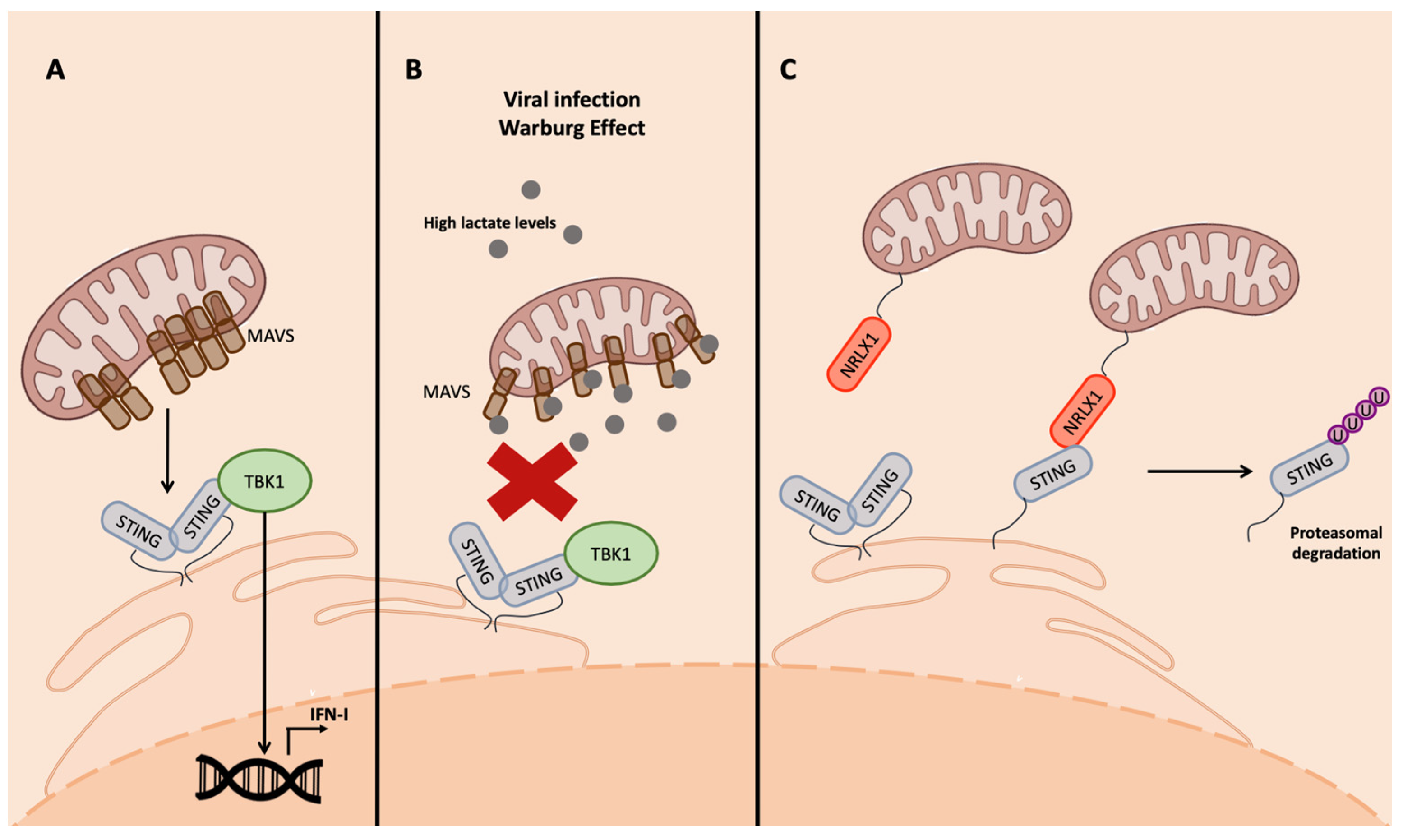
4. STING Connection with Mitochondria
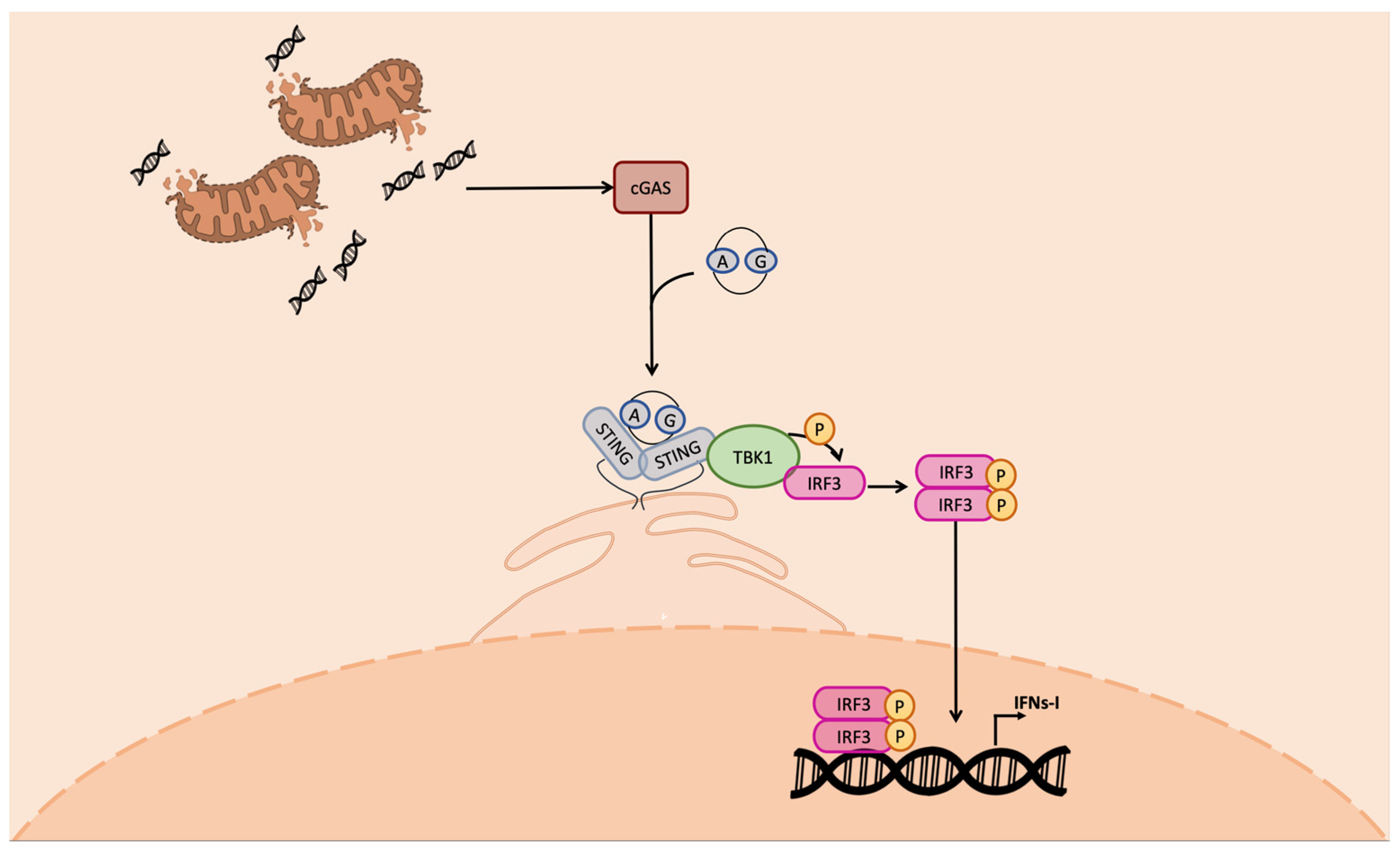
5. Triggering of STING through mtDNA Release
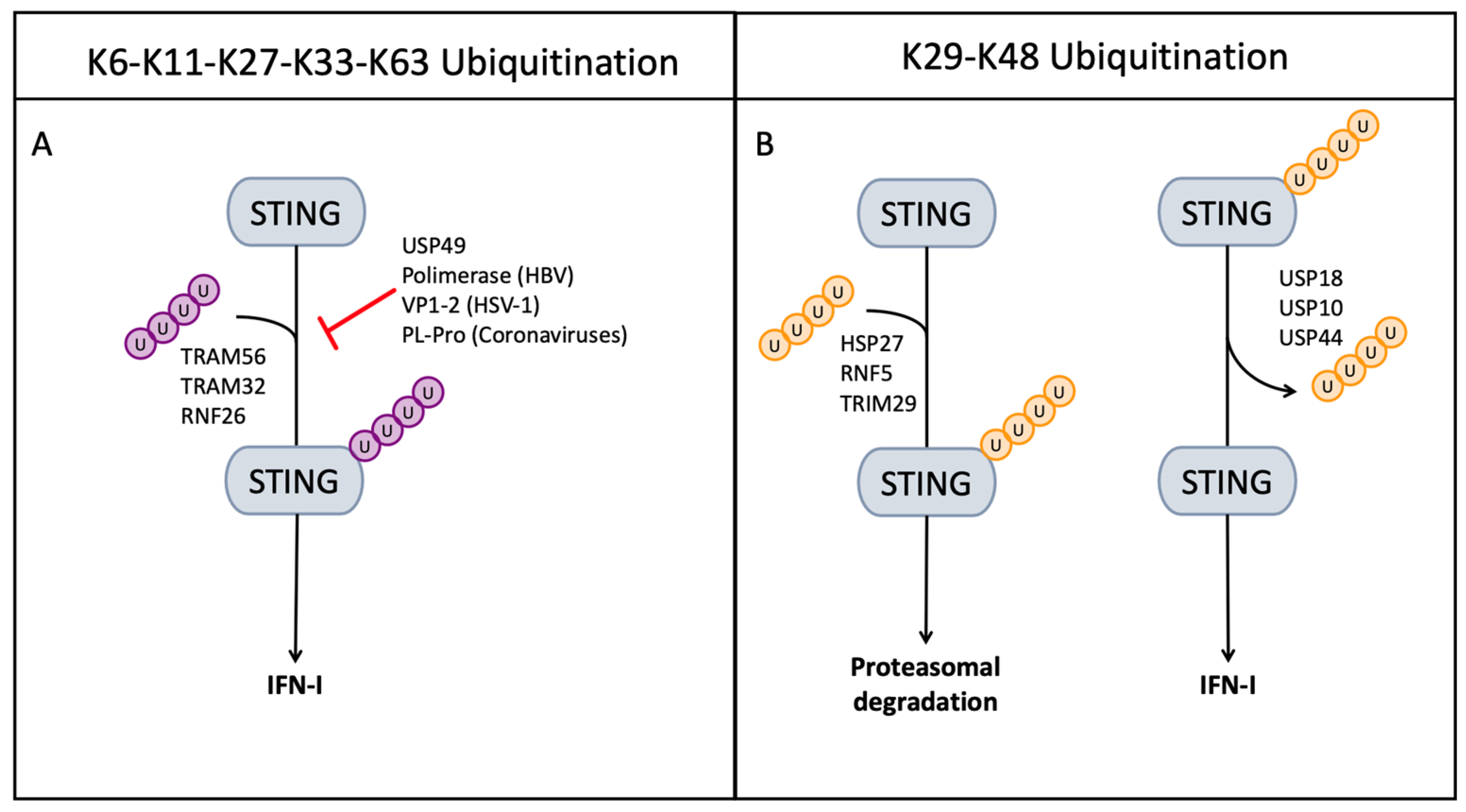
6. Role of Ubiquitin in the STING-Mediated Innate Immune Response
7. Direct Viral STING Inhibition
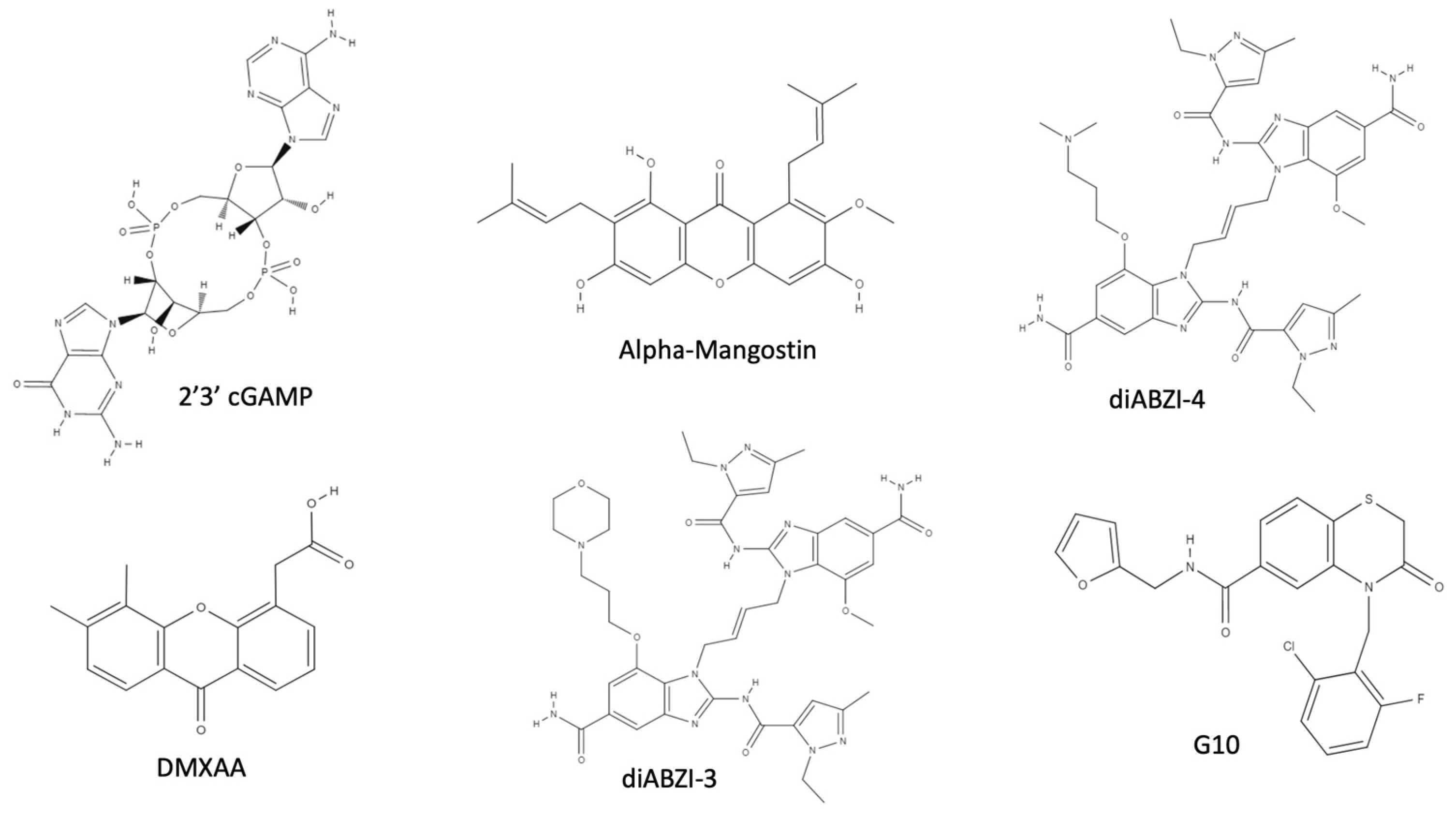
8. STING Agonists as Broad-Spectrum Antivirals
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gallo, R.L.; Nizet, V. Innate barriers against infection and associated disorders. Drug. Discov. Today Dis. Mech. 2008, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Crozat, K.; Vivier, E.; Dalod, M. Crosstalk between components of the innate immune system: Promoting anti-microbial defenses and avoiding immunopathologies. Immunol. Rev. 2009, 227, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Signaling pathways activated by microorganisms. Curr. Opin. Cell Biol. 2007, 19, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Itav, S.; Elinav, E. Integration of Innate Immune Signaling. Trends Immunol. 2016, 37, 84–101. [Google Scholar] [CrossRef]
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.H.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug. Discov. 2016, 15, 35–50. [Google Scholar] [CrossRef]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.R.; Prokunina-Olsson, L.; Donnelly, R.P. IFN-λ4: The Paradoxical New Member of the Interferon Lambda Family. J. Interferon Cytokine Res. 2014, 34, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Perelson, A.S. Treatment of Hepatitis C Virus Infection with Interferon and Small Molecule Direct Antivirals: Viral Kinetics and Modeling. Crit. Rev. Immunol. 2010, 30, 131–148. [Google Scholar] [CrossRef]
- Silva, M.O. Risk of autoimmune complications associated with interferon therapy. Gastroenterol. Hepatol. 2012, 8, 540–542. [Google Scholar]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.M.; Edwards, M.R.; Diederichs, A.; Alinger, J.B.; Leung, D.W.; Amarasinghe, G.K.; Basler, C.F. VP24-Karyopherin Alpha Binding Affinities Differ between Ebolavirus Species, Influencing Interferon Inhibition and VP24 Stability. J. Virol. 2017, 91, e01715-16. [Google Scholar] [CrossRef]
- Shelemba, A.A.; Lushnikova, E.L.; Kolesnikov, S.I.; Nepomnyashchikh, L.M.; Chepurnov, A.A. Role of Ebola Virus vp24 Protein in Inhibition of Interferonogenesis. Bull. Exp. Biol. Med. 2016, 160, 350–352. [Google Scholar] [CrossRef]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and Interferon Antagonism Activities of Coronavirus Papain-Like Proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X.; Bian, G.; Tu, J.; Xing, Y.; Wang, Y.; Chen, Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014, 95, 614–626. [Google Scholar] [CrossRef]
- Mielech, A.M.; Chen, Y.; Mesecar, A.D.; Baker, S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014, 194, 184–190. [Google Scholar] [CrossRef]
- Pronin, A.V.; Narovlyansky, A.N.; Sanin, A.V. New Approaches to the Prevention and Treatment of Viral Diseases. Arch. Immunol. Ther. Exp. (Warsz) 2021, 69, 10. [Google Scholar] [CrossRef] [PubMed]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Es-Saad, S.; Tremblay, N.; Baril, M.; Lamarre, D. Regulators of innate immunity as novel targets for panviral therapeutics. Curr. Opin. Virol. 2012, 2, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Cordova, A.F.; Li, L. Therapeutic Interventions Targeting Innate Immune Receptors: A Balancing Act. Chem. Rev. 2022, 122, 3414–3458. [Google Scholar] [CrossRef] [PubMed]
- Pattabhi, S.; Wilkins, C.R.; Dong, R.; Knoll, M.L.; Posakony, J.; Kaiser, S.; Mire, C.E.; Wang, M.L.; Ireton, R.C.; Geisbert, T.W.; et al. Targeting Innate Immunity for Antiviral Therapy through Small Molecule Agonists of the RLR Pathway. J. Virol. 2016, 90, 2372–2387. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor Activity of a Systemic STING-Activating Non-Nucleotide cGAMP Mimetic. Available online: http://science.sciencemag.org/ (accessed on 1 March 2023).
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Berger, G.; Lawler, S.E. Novel non-nucleotidic STING agonists for cancer immunotherapy. Future Med. Chem. 2018, 10, 2767–2769. [Google Scholar] [CrossRef]
- Corrales, L.; Gajewski, T.F. Endogenous and pharmacologic targeting of the STING pathway in cancer immunotherapy. Cytokine 2016, 77, 245–247. [Google Scholar] [CrossRef]
- Guo, F.; Han, Y.; Zhao, X.; Wang, J.; Liu, F.; Xu, C.; Wei, L.; Jiang, J.-D.; Block, T.M.; Guo, J.-T.; et al. STING Agonists Induce an Innate Antiviral Immune Response against Hepatitis B Virus. Antimicrob. Agents Chemother. 2015, 59, 1273–1281. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Maringer, K.; Fernandez-Sesma, A. Message in a bottle: Lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor. Rev. 2014, 25, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658. [Google Scholar] [CrossRef]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Deimling, T.; Mann, C.C.D.O.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Gao, P.; Gao, G.; Fan, Z. DNA sensor cGAS-mediated immune recognition. Protein Cell 2016, 7, 777–791. [Google Scholar] [CrossRef]
- Tamayo, R.; Pratt, J.T.; Camilli, A. Roles of Cyclic Diguanylate in the Regulation of Bacterial Pathogenesis. Annu. Rev. Microbiol. 2007, 61, 131–148. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe 2015, 17, 799–810. [Google Scholar] [CrossRef]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science (1979) 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Shu, H.-B.; Wang, Y.-Y. MITA/STING: A central and multifaceted mediator in innate immune response. Cytokine Growth Factor. Rev. 2014, 25, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Wen, Y.; Shu, C.; Han, Q.; Konan, K.V.; Li, P.; Kao, C.C. Hepatitis C Virus NS4B Can Suppress STING Accumulation to Evade Innate Immune Responses. J. Virol. 2016, 90, 254–265. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef]
- Unterholzner, L.; Dunphy, G. cGAS-independent STING activation in response to DNA damage. Mol. Cell. Oncol. 2019, 6, 1558682. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e5. [Google Scholar] [CrossRef]
- Hussain, B.; Xie, Y.; Jabeen, U.; Lu, D.; Yang, B.; Wu, C.; Shang, G. Activation of STING Based on Its Structural Features. Front. Immunol. 2022, 13, 808607. [Google Scholar] [CrossRef]
- Ouyang, S.; Song, X.; Wang, Y.; Ru, H.; Shaw, N.; Jiang, Y.; Niu, F.; Zhu, Y.; Qiu, W.; Parvatiyar, K.; et al. Structural Analysis of the STING Adaptor Protein Reveals a Hydrophobic Dimer Interface and Mode of Cyclic di-GMP Binding. Immunity 2012, 36, 1073–1086. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins Struct. Funct. Genetics 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer, v20.1.0.19295; Dassault Systèmes: San Diego, Brazil, 2020. [Google Scholar]
- Shang, G.; Zhu, D.; Li, N.; Zhang, J.; Zhu, C.; Lu, D.; Liu, C.; Yu, Q.; Zhao, Y.; Xu, S.; et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 2012, 19, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.; Yi, G.; Watts, T.; Kao, C.C.; Li, P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 2012, 19, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-Function Analysis of STING Activation by c[G(2′,5′)pA(3′,5′)p] and Targeting by Antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef]
- Cong, X.; Yuan, Z.; Du, Y.; Wu, B.; Lu, D.; Wu, X.; Zhang, Y.; Li, F.; Wei, B.; Li, J.; et al. Crystal structures of porcine STINGCBD–CDN complexes reveal the mechanism of ligand recognition and discrimination of STING proteins. J. Biol. Chem. 2019, 294, 11420–11432. [Google Scholar] [CrossRef]
- Decker, T.; Müller, M.; Stockinger, S. The Yin and Yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci. Rep. 2016, 6, srep19049. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Bahat, A.; MacVicar, T.; Langer, T. Metabolism and Innate Immunity Meet at the Mitochondria. Front. Cell Dev. Biol. 2021, 9, 720490. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, G.; Xu, Z.-G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, R.; Fang, P.; Li, M.; Yu, H.; Wang, Q.; Yu, Y.; Wang, F.; Zhang, Y.; Chen, A.; et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat. Commun. 2021, 12, 98. [Google Scholar] [CrossRef]
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F.V. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9035–9040. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Pickering, R.J.; Booty, L.M. NLR in eXile: Emerging roles of NLRX1 in immunity and human disease. Immunology 2021, 162, 268–280. [Google Scholar] [CrossRef]
- Nagai-Singer, M.A.; Morrison, H.A.; Allen, I.C. NLRX1 Is a Multifaceted and Enigmatic Regulator of Immune System Function. Front. Immunol. 2019, 10, 2419. [Google Scholar] [CrossRef]
- Guo, H.; König, R.; Deng, M.; Rieß, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef]
- Luo, X.; Donnelly, C.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N. Functional organization of mammalian mitochondrial DNA in nucleoids: History, recent developments, and future challenges. IUBMB Life 2009, 62, 19–32. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef]
- Lucas-Hourani, M.; Dauzonne, D.; Munier-Lehmann, H.; Khiar, S.; Nisole, S.; Dairou, J.; Helynck, O.; Afonso, P.V.; Tangy, F.; Vidalain, P.-O. Original Chemical Series of Pyrimidine Biosynthesis Inhibitors That Boost the Antiviral Interferon Response. Antimicrob. Agents Chemother. 2017, 61, e00383-17. [Google Scholar] [CrossRef]
- Lucas-Hourani, M.; Dauzonne, D.; Jorda, P.; Cousin, G.; Lupan, A.; Helynck, O.; Caignard, G.; Janvier, G.; André-Leroux, G.; Khiar, S.; et al. Inhibition of Pyrimidine Biosynthesis Pathway Suppresses Viral Growth through Innate Immunity. PLoS Pathog. 2013, 9, e1003678. [Google Scholar] [CrossRef]
- Hayek, S.; Pietrancosta, N.; Hovhannisyan, A.A.; de Sousa, R.A.; Bekaddour, N.; Ermellino, L.; Tramontano, E.; Arnould, S.; Sardet, C.; Dairou, J.; et al. Cerpegin-derived furo [3,4-c]pyridine-3,4(1H,5H)-diones enhance cellular response to interferons by de novo pyrimidine biosynthesis inhibition. Eur. J. Med. Chem. 2020, 186, 111855. [Google Scholar] [CrossRef]
- Liu, Q.; Gupta, A.; Okesli-Armlovich, A.; Qiao, W.; Fischer, C.R.; Smith, M.; Carette, J.E.; Bassik, M.C.; Khosla, C. Enhancing the Antiviral Efficacy of RNA-Dependent RNA Polymerase Inhibition by Combination with Modulators of Pyrimidine Metabolism. Cell Chem. Biol. 2020, 27, 668–677.e9. [Google Scholar] [CrossRef]
- Cheung, N.N.; Lai, K.K.; Dai, J.; Kok, K.H.; Chen, H.; Chan, K.-H.; Yuen, K.-Y.; Kao, R.Y.T. Broad-spectrum inhibition of common respiratory RNA viruses by a pyrimidine synthesis inhibitor with involvement of the host antiviral response. J. Gen. Virol. 2017, 98, 946–954. [Google Scholar] [CrossRef]
- Xiong, R.; Zhang, L.; Li, S.; Sun, Y.; Ding, M.; Wang, Y.; Zhao, Y.; Wu, Y.; Shang, W.; Jiang, X.; et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell 2020, 11, 723–739. [Google Scholar] [CrossRef]
- Okesli, A.; Khosla, C.; Bassik, M.C. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr. Opin. Biotechnol. 2017, 48, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.-H.; Kunz, A.; Simon, V.A.; Palese, P.; Shaw, M.L. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Kim, C.; Cho, S. Gemcitabine and Nucleos(t)ide Synthesis Inhibitors Are Broad-Spectrum Antiviral Drugs that Activate Innate Immunity. Viruses 2018, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The Ubiquitin-Proteasome Pathway and Proteasome Inhibitors’. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.C. Ubiquitin signaling in immune responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef]
- Schlesinger, D.H.; Goldstein, G.; Niall, H.D. Complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells. Biochemistry 1975, 14, 2214–2218. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IκB Kinase Complex by TRAF6 Requires a Dimeric Ubiquitin-Conjugating Enzyme Complex and a Unique Polyubiquitin Chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Jin, L.; Williamson, A.; Banerjee, S.; Philipp, I.; Rape, M. Mechanism of Ubiquitin-Chain Formation by the Human Anaphase-Promoting Complex. Cell 2008, 133, 653–665. [Google Scholar] [CrossRef]
- Talis, A.L.; Huibregtse, J.M.; Howley, P.M. The Role of E6AP in the Regulation of p53 Protein Levels in Human Papillomavirus (HPV)-positive and HPV-negative Cells. J. Biol. Chem. 1998, 273, 6439–6445. [Google Scholar] [CrossRef]
- Monia, B.P.; Ecker, D.J.; Jonnalagadda, S.; Marsh, J.; Gotlib, L.; Butt, T.R.; Crooke, S.T. Gene Synthesis, Expression, and Processing of Human Ubiquitin Carboxyl Extension Proteins. J. Biol. Chem. 1989, 264, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- Saferding, V.; Blüml, S. Innate immunity as the trigger of systemic autoimmune diseases. J. Autoimmun. 2020, 110, 102382. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Ma, T.; Liu, X.; Wang, C. cGAS-STING pathway: Post-translational modifications and functions in sterile inflammatory diseases. FEBS J. 2022, 289, 6187–6208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhou, M.-T.; Hu, M.-M.; Hu, Y.-H.; Zhang, J.; Guo, L.; Zhong, B.; Shu, H.-B. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PLoS Pathog. 2014, 10, e1004358. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.-P.; Yang, Y.; Wang, Y.-Y.; Zhang, X.-L.; Shu, H.-B. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef]
- Li, X.; Xie, J.; Li, D.; Li, H.; Niu, Y.; Wu, B.; Yang, Y.; Yan, Z.; Zhang, X.; Chen, L.; et al. HSP27 Attenuates cGAS-Mediated IFN-β Signaling through Ubiquitination of cGAS and Promotes PRV Infection. Viruses 2022, 14, 1851. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.-X.; Zhang, Q.; Zhu, G.-F.; Yuan, L.; Zhang, D.-E.; Zhu, Q.; Yao, J.; Shu, H.-B.; Zhong, B. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016, 26, 1302–1319. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, Q.; Liuyu, T.; Xu, Z.; Zhang, M.-X.; Luo, M.-H.; Zeng, W.-B.; Zhu, Q.; Lin, D.; Zhong, B. USP49 negatively regulates cellular antiviral responses via deconjugating K63-linked ubiquitination of MITA. PLoS Pathog. 2019, 15, e1007680. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y. Potential interventions for novel coronavirus in China: A systematic review. J. Med. Virol. 2020, 92, 479–490. [Google Scholar] [CrossRef]
- Karhausen, J.; Ulloa, L.; Yang, W. SUMOylation Connects Cell Stress Responses and Inflammatory Control: Lessons from the Gut as a Model Organ. Front. Immunol. 2021, 12, 646633. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-M.; Yang, Q.; Xie, X.-Q.; Liao, C.-Y.; Lin, H.; Liu, T.-T.; Yin, L.; Shu, H.-B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING to Block Innate Cytosolic DNA-Sensing Pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jiang, L.; Chen, S.; Hu, Q.; Huang, Y.; Wu, Y.; Chen, W. HBx inhibits DNA sensing signaling pathway via ubiquitination and autophagy of cGAS. Virol. J. 2022, 19, 55. [Google Scholar] [CrossRef]
- Bodda, C.; Reinert, L.S.; Fruhwürth, S.; Richardo, T.; Sun, C.; Zhang, B.-C.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergström, P.; et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217, e20191422. [Google Scholar] [CrossRef]
- Sun, L.; Xing, Y.; Chen, X.; Zheng, Y.; Yang, Y.; Nichols, D.B.; Clementz, M.A.; Banach, B.S.; Li, K.; Baker, S.C.; et al. Coronavirus Papain-like Proteases Negatively Regulate Antiviral Innate Immune Response through Disruption of STING-Mediated Signaling. PLoS ONE 2012, 7, e30802. [Google Scholar] [CrossRef]
- Clemente, V.; D’Arcy, P.; Bazzaro, M. Deubiquitinating Enzymes in Coronaviruses and Possible Therapeutic Opportunities for COVID-19. Int. J. Mol. Sci. 2020, 21, 3492. [Google Scholar] [CrossRef]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP 27 targets the TBK 1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-Sensing Pathway by VP11/12 of Herpes Simplex Virus 1. J. Virol. 2017, 91, e00535-17. [Google Scholar] [CrossRef]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 γ 1 34.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Su, S.; Gao, Y.-Q.; Wang, P.-P.; Huang, Z.-F.; Hu, M.-M.; Luo, W.-W.; Li, S.; Luo, M.-H.; Wang, Y.-Y.; et al. Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Guo, Y.; Zou, H.-M.; Su, S.; Wang, S.-Y.; Yang, Q.; Luo, M.-H.; Wang, Y.-Y. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog. 2019, 15, e1007691. [Google Scholar] [CrossRef]
- Kim, J.-E.; Kim, Y.-E.; Stinski, M.F.; Ahn, J.-H.; Song, Y.-J. Human Cytomegalovirus IE2 86 kDa Protein Induces STING Degradation and Inhibits cGAMP-Mediated IFN-β Induction. Front. Microbiol. 2017, 8, 1854. [Google Scholar] [CrossRef]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.-J.; Kato, N.; Shu, H.-B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Su, M.; Zheng, J.; Gan, L.; Zhao, Y.; Fu, Y.; Chen, Q. Second messenger 2′3′-cyclic GMP–AMP (2′3′-cGAMP): Synthesis, transmission, and degradation. Biochem. Pharmacol. 2022, 198, 114934. [Google Scholar] [CrossRef]
- Skouboe, M.K.; Knudsen, A.; Reinert, L.; Boularan, C.; Lioux, T.; Pérouzel, E.; Thomsen, M.K.; Paludan, S.R. STING agonists enable antiviral cross-talk between human cells and confer protection against genital herpes in mice. PLoS Pathog. 2018, 14, e1006976. [Google Scholar] [CrossRef]
- Cai, H.; Holleufer, A.; Simonsen, B.; Schneider, J.; Lemoine, A.; Gad, H.H.; Huang, J.; Huang, J.; Chen, D.; Peng, T.; et al. 2′3′-cGAMP triggers a STING- and NF-κB–dependent broad antiviral response in Drosophila. Sci. Signal. 2020, 13, eabc4537. [Google Scholar] [CrossRef]
- Liu, B.; Tang, L.; Zhang, X.; Ma, J.; Sehgal, M.; Cheng, J.; Zhang, X.; Zhou, Y.; Du, Y.; Kulp, J.; et al. A cell-based high throughput screening assay for the discovery of cGAS-STING pathway agonists. Antiviral Res. 2017, 147, 37–46. [Google Scholar] [CrossRef]
- Jameson, M.B.; on behalf of the Phase I/II Trials Committee of Cancer Research UK; Thompson, P.I.; Baguley, B.C.; Evans, B.D.; Harvey, V.J.; Porter, D.J.; McCrystal, M.R.; Small, M.; Bellenger, K.; et al. Clinical aspects of a phase I trial of 5,6-dimethylxanthenone-4-acetic acid (DMXAA), a novel antivascular agent. Br. J. Cancer 2003, 88, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Li, L.; Maliga, Z.; Yin, Q.; Wu, H.; Mitchison, T.J. Anticancer Flavonoids Are Mouse-Selective STING Agonists. ACS Chem. Biol. 2013, 8, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zillinger, T.; Wang, W.; Ascano, M.; Dai, P.; Hartmann, G.; Tuschl, T.; Deng, L.; Barchet, W.; Patel, D.J. Binding-Pocket and Lid-Region Substitutions Render Human STING Sensitive to the Species-Specific Drug DMXAA. Cell Rep. 2014, 8, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Tarasuk, M.; Songprakhon, P.; Chieochansin, T.; Choomee, K.; Na-Bangchang, K.; Yenchitsomanus, P. Alpha-mangostin inhibits viral replication and suppresses nuclear factor kappa B (NF-κB)-mediated inflammation in dengue virus infection. Sci. Rep. 2022, 12, 16088. [Google Scholar] [CrossRef]
- Yongpitakwattana, P.; Morchang, A.; Panya, A.; Sawasdee, N.; Yenchitsomanus, P. Alpha-mangostin inhibits dengue virus production and pro-inflammatory cytokine/chemokine expression in dendritic cells. Arch. Virol. 2021, 166, 1623–1632. [Google Scholar] [CrossRef]
- Choi, M.; Kim, Y.-M.; Lee, S.; Chin, Y.-W.; Lee, C. Mangosteen xanthones suppress hepatitis C virus genome replication. Virus Genes 2014, 49, 208–222. [Google Scholar] [CrossRef]
- Panda, K.; Alagarasu, K.; Patil, P.; Agrawal, M.; More, A.; Kumar, N.; Mainkar, P.; Parashar, D.; Cherian, S. In Vitro Antiviral Activity of α-Mangostin against Dengue Virus Serotype-2 (DENV-2). Molecules 2021, 26, 3016. [Google Scholar] [CrossRef]
- Cavlar, T.; Deimling, T.; Ablasser, A.; Hopfner, K.-P.; Hornung, V. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J. 2013, 32, 1440–1450. [Google Scholar] [CrossRef]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.-Y.; Tran, J.-L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef]
- Zhu, Q.; Hu, H.; Liu, H.; Shen, H.; Yan, Z.; Gao, L. A synthetic STING agonist inhibits the replication of human parainfluenza virus 3 and rhinovirus 16 through distinct mechanisms. Antivir. Res. 2020, 183, 104933. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, Y.; Wang, L.; Yao, X.; Wu, D.; Cheng, J.; Pan, X.; Liu, H.; Yan, Z.; Gao, L. Inhibition of coronavirus infection by a synthetic STING agonist in primary human airway system. Antivir. Res. 2021, 187, 105015. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Reyes, H.M.; Yang, J.F.; Li, Y.; Stewart, K.M.; Basil, M.C.; Lin, S.M.; Katzen, J.; Morrisey, E.E.; Weiss, S.R.; et al. Activation of STING Signaling Pathway Effectively Blocks Human Coronavirus Infection. J. Virol. 2021, 95, e00490-21. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ferretti, M.; Ying, B.; Descamps, H.; Lee, E.; Dittmar, M.; Lee, J.S.; Whig, K.; Kamalia, B.; Dohnalová, L.; et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabi9007. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Shmuel-Galia, L.; Jiang, Z.; Wilson, R.; Landis, P.; Ng, S.-L.; Parsi, K.M.; Maehr, R.; Cruz, J.; Morales-Ramos, A.; et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Sci. Immunol. 2021, 6, abi9002. [Google Scholar] [CrossRef] [PubMed]
- Sali, T.M.; Pryke, K.M.; Abraham, J.; Liu, A.; Archer, I.; Broeckel, R.; Staverosky, J.A.; Smith, J.L.; Al-Shammari, A.; Amsler, L.; et al. Characterization of a Novel Human-Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015, 11, e1005324. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating Activity of SARS-CoV Papain-Like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Cheng, W.; Chen, S.; Li, R.; Chen, Y.; Wang, M.; Guo, D. Severe acute respiratory syndrome coronavirus protein 6 mediates ubiquitin-dependent proteosomal degradation of N-Myc (and STAT) interactor. Virol. Sin. 2015, 30, 153–161. [Google Scholar] [CrossRef]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus Papain-Like Protease Ubiquitin-Like Domain and Catalytic Domain Regulate Antagonism of IRF3 and NF-κB Signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef]
- Soh, S.-M.; Kim, Y.-J.; Kim, H.-H.; Lee, H.-R. Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses. Int. J. Mol. Sci. 2022, 23, 492. [Google Scholar] [CrossRef]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.M.; Wolf, L.M.; Bello, G.L.; Maccari, J.G.; Nasi, L.A. Molecular evolution of SARS-CoV-2 from December 2019 to August 2022. J. Med. Virol. 2023, 95, e28366. [Google Scholar] [CrossRef] [PubMed]
- Guerini, D. STING Agonists/Antagonists: Their Potential as Therapeutics and Future Developments. Cells 2022, 11, 1159. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Pei, J.; Luo, Q.; Zeng, X.; Li, Q.; Yang, Z.; Quan, J. Identification of α-Mangostin as an Agonist of Human STING. ChemMedChem 2018, 13, 2057–2064. [Google Scholar] [CrossRef] [PubMed]
- Margolis, S.R.; Dietzen, P.A.; Hayes, B.M.; Wilson, S.C.; Remick, B.C.; Chou, S.; Vance, R.E. The cyclic dinucleotide 2′3′-cGAMP induces a broad antibacterial and antiviral response in the sea anemone Nematostella vectensis. Proc. Natl. Acad. Sci. USA 2021, 118, e2109022118. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulis, A.; Tramontano, E. Unlocking STING as a Therapeutic Antiviral Strategy. Int. J. Mol. Sci. 2023, 24, 7448. https://doi.org/10.3390/ijms24087448
Paulis A, Tramontano E. Unlocking STING as a Therapeutic Antiviral Strategy. International Journal of Molecular Sciences. 2023; 24(8):7448. https://doi.org/10.3390/ijms24087448
Chicago/Turabian StylePaulis, Annalaura, and Enzo Tramontano. 2023. "Unlocking STING as a Therapeutic Antiviral Strategy" International Journal of Molecular Sciences 24, no. 8: 7448. https://doi.org/10.3390/ijms24087448