
Structural and Pathogenic Impacts of ABCA4 Variants in Retinal Degenerations—An In-Silico Study

Abstract
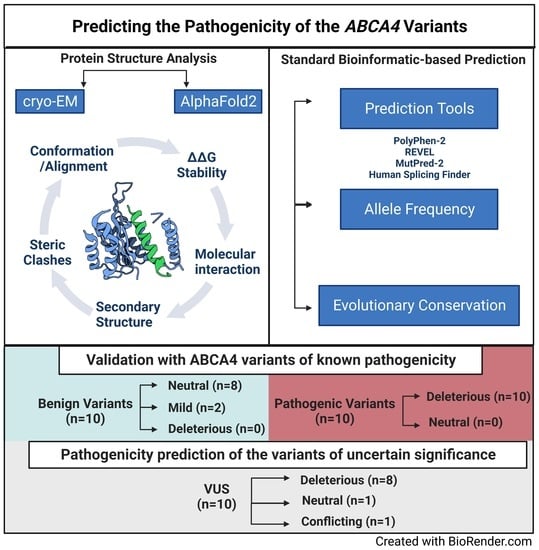
:
1. Introduction
2. Results
2.1. AlphaFold2 Protein Modeling
2.2. In Silico Protein Structural Analysis and Pathogenicity Prediction of the ABCA4 Variants of Known Significance
2.2.1. In Silico Analysis of ABCA4 Benign Variants
2.2.2. In Silico Analysis of ABCA4 Pathogenic Variants
2.3. In Silico Pipeline Analysis of ABCA4 Variants of Uncertain Significance
3. Discussion
4. Materials and Methods
4.1. Curation of the ABCA4 Variants from Databases and Pathogenicity Prediction
4.2. AlphaFold2 Protein Modeling
4.3. Protein Structure Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Nathans, J. Mechanistic studies of ABCR, the ABC transporter in photoreceptor outer segments responsible for autosomal recessive Stargardt disease. J. Bioenerg. Biomembr. 2001, 33, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A. Historical evolution in the understanding of Stargardt macular dystrophy. Ophthalmic Genet. 2010, 31, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Starqardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Azarian, S.M.; Travis, G.H. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt’s disease (ABCR). FEBS Lett. 1997, 409, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Nathans, J. [58] ABCR: Rod photoreceptor-specific ABC transporter responsible for Stargardt disease. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2000; Volume 315, pp. 879–897. [Google Scholar]
- Koenekoop, R.K. The gene for Stargardt disease, ABCA4, is a major retinal gene: A mini-review. Ophthalmic Genet. 2003, 24, 75–80. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; van Driel, M.; den Hollander, A.I.; van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef] [Green Version]
- Klevering, B.J.; Yzer, S.; Rohrschneider, K.; Zonneveld, M.; Allikmets, R.; van den Born, L.I.; Maugeri, A.; Hoyng, C.B.; Cremers, F.P. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur. J. Hum. Genet. 2004, 12, 1024–1032. [Google Scholar] [CrossRef]
- Martínez-Mir, A.; Paloma, E.; Allikmets, R.; Ayuso, C.; del Rio, T.; Dean, M.; Vilageliu, L.; Gonzàlez-Duarte, R.; Balcells, S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet. 1998, 18, 11–12. [Google Scholar] [CrossRef]
- Allikmets, R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am. J. Hum. Genet. 2000, 67, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Dean, M. Bringing age-related macular degeneration into focus. Nat. Genet. 2008, 40, 820–821. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fleckenstein, M.; Fiebig, B.S.; Schmitz-Valckenberg, S.; Bindewald-Wittich, A.; Keilhauer, C.N.; Renner, A.B.; Mackensen, F.; Mößner, A.; Pauleikhoff, D.; et al. A subgroup of age-related macular degeneration is associated with mono-allelic sequence variants in the ABCA4 gene. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2112–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, A.; White, K.; Stöhr, H.; Steiner, K.; Hemmrich, N.; Grimm, T.; Jurklies, B.; Lorenz, B.; Scholl, H.P.; Apfelstedt-Sylla, E.; et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 2000, 67, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Tian, L.; Huang, Y. Correlation between the interactions of ABCA4 polymorphisms and smoking with the susceptibility to age-related macular degeneration. Int. J. Clin. Exp. Pathol. 2015, 8, 7403–7408. [Google Scholar] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb. Mol. Case Stud. 2018, 4, 2733. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Smallwood, P.M.; Nathans, J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat. Genet. 2000, 26, 242–246. [Google Scholar] [CrossRef]
- Biswas-Fiss, E.E. Functional analysis of genetic mutations in nucleotide binding domain 2 of the human retina specific ABC transporter. Biochemistry 2003, 42, 10683–10696. [Google Scholar] [CrossRef]
- Biswas-Fiss, E.E.; Affet, S.; Ha, M.; Biswas, S.B. Retinoid binding properties of nucleotide binding domain 1 of the Stargardt disease-associated ATP binding cassette (ABC) transporter, ABCA4. J. Biol. Chem. 2012, 287, 44097–44107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.J.; Biswas, S.B.; Biswas-Fiss, E.E. Functional significance of the conserved C-Terminal VFVNFA motif in the retina-specific ABC transporter, ABCA4, and its role in inherited visual disease. Biochem. Biophys. Res. Commun. 2019, 519, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Garces, F.; Jiang, K.; Molday, L.L.; Stöhr, H.; Weber, B.H.; Lyons, C.J.; Maberley, D.; Molday, R.S. Correlating the Expression and Functional Activity of ABCA4 Disease Variants with the Phenotype of Patients with Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2305–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garces, F.A.; Scortecci, J.F.; Molday, R.S. Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration. Int. J. Mol.Sci. 2020, 22, 185. [Google Scholar] [CrossRef]
- Curtis, S.B.; Molday, L.L.; Garces, F.A.; Molday, R.S. Functional analysis and classification of homozygous and hypomorphic ABCA4 variants associated with Stargardt macular degeneration. Hum. Mutat. 2020, 41, 1944–1956. [Google Scholar] [CrossRef]
- Wiszniewski, W.; Zaremba, C.M.; Yatsenko, A.N.; Jamrich, M.; Wensel, T.G.; Lewis, R.A.; Lupski, J.R. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum. Mol. Genet. 2005, 14, 2769–2778. [Google Scholar] [CrossRef] [Green Version]
- Biswas-Fiss, E.E.; Kurpad, D.S.; Joshi, K.; Biswas, S.B. Interaction of extracellular domain 2 of the human retina-specific ATP-binding cassette transporter (ABCA4) with all-trans-retinal. J. Biol. Chem. 2010, 285, 19372–19383. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39, W215–W222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Capriotti, E.; Altman, R.B. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinform. 2011, 12 (Suppl. 4), S3. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lee, J.; Chen, J. Molecular structures of the eukaryotic retinal importer ABCA4. eLife 2021, 10, e63524. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, Z.; Fang, Q.; Du, B.; Gong, X. Structural basis of substrate recognition and translocation by human ABCA4. Nat. Commun. 2021, 12, 3853. [Google Scholar] [CrossRef] [PubMed]
- Scortecci, J.F.; Molday, L.L.; Curtis, S.B.; Garces, F.A.; Panwar, P.; Van Petegem, F.; Molday, R.S. Cryo-EM structures of the ABCA4 importer reveal mechanisms underlying substrate binding and Stargardt disease. Nat. Commun. 2021, 12, 5902. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View-molecular graphics for all devices-from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lee, J.; Chen, J. ATP-free human ABCA4. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.; Chen, J. ATP-bound human ABCA4. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, Z.K.; Gong, X. Human ABCA4 in the apo state. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, Z.K.; Gong, X. Human ABCA4 in NRPE-bound state. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, Z.K.; Gong, X. Human ABCA4 in ATP-bound state. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Scortecci, J.F.; Van Petegem, F.; Molday, R.S. Human ABCA4 structure in the unbound state. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Scortecci, J.F.; Van Petegem, F.; Molday, R.S. Human ABCA4 structure in complex with N-ret-PE. Worldw. Protein Data Bank 2021. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In Silico Functional Meta-Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Human Mutation 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Qu, L.H.; Jin, X.; Zeng, C.; Zhou, N.G.; Liu, Y.H.; Lin, Y. Targeted next-generation sequencing identifies ABCA4 mutations in Chinese families with childhood-onset and adult-onset Stargardt disease. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T. Structural dynamics of ABC transporters: Molecular simulation studies. Biochem. Soc. Trans. 2021, 49, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Hoffmann, B.; Lehn, P.; Mornon, J.-P. Molecular modelling and molecular dynamics of CFTR. Cell. Mol. Life Sci. 2017, 74, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, K.P.; Gao, X.; Zhao, J.; Kim, K.-S.; Maloney, S.E.; Gotoff, J.; Parikh, S.; Leu, Y.-C.; Wu, K.-P.; Shinawi, M.; et al. Identification of disease-linked hyperactivating mutations in UBE3A through large-scale functional variant analysis. Nat. Commun. 2021, 12, 6809. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglio, P.I.; Bruque, C.D.; Lotersztein, V.; Luce, L.; Giliberto, F.; Menazzi, S.; Francipane, L.; Paoli, B.; Goldschmidt, E.; Elgoyhen, A.B.; et al. Predicting pathogenicity for novel hearing loss mutations based on genetic and protein structure approaches. Sci. Rep. 2022, 12, 301. [Google Scholar] [CrossRef]
- Li, B.; Yang, Y.T.; Capra, J.A.; Gerstein, M.B. Predicting changes in protein thermodynamic stability upon point mutation with deep 3D convolutional neural networks. PLoS Comput. Biol. 2020, 16, e1008291. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [Green Version]
- Suybeng, V.; Koeppel, F.; Harlé, A.; Rouleau, E. Comparison of Pathogenicity Prediction Tools on Somatic Variants. J. Mol. Diagn. 2020, 22, 1383–1392. [Google Scholar] [CrossRef]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef]
- Khabou, B.; Durand-Schneider, A.-M.; Delaunay, J.-L.; Aït-Slimane, T.; Barbu, V.; Fakhfakh, F.; Housset, C.; Maurice, M. Comparison of in silico prediction and experimental assessment of ABCB4 variants identified in patients with biliary diseases. Int. J. Biochem. Cell Biol. 2017, 89, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Significance | ABCA4 Variant | Previous Functional Studies | Allele Freq. | Predicted Pathogenicity (Collective) | RMSD (Å) | TM-Score | In Silico ∆∆G (kcal/mol) | Predicted Effect on Structure |
|---|---|---|---|---|---|---|---|---|
| Benign | c.229G > A(p.V77M) | - | - | Benign | 0.534 | 0.9333 | −0.09 | Neu |
| c.635G > A(p.R212H) | -(R212C: ↓ ATPase [26]) | 4.24 × 10−2 | Pathogenic a | 0.793 | 0.9372 | −5.19 | Neu | |
| c.1268A > G(p.H423R) | - | 2.56 × 10−1 | Benign | 0.647 | 0.9333 | −2.3 | Mild | |
| c.3626T > C(p.M1209T) | - | 3.03 × 10−3 | Benign | 0.539 | 0.9034 | +0.71 | Neu | |
| c.3899G > A(p.R1300Q) | - | 6.70 × 10−3 | Benign | 0.775 | 0.9507 | +0.007 | Neu | |
| c.4283C > T(p.T1428M) | - | 4.44 × 10−3 | Benign | 0.660 | 0.9865 | −0.85 | Neu | |
| c.4503G > C(p.E1501D) | - | 1.12 × 10−3 | Benign | 0.326 | 0.9199 | −0.38 | Mild | |
| c.5843_5844inv (p.P1948L) | - | 3.14 × 10−2 | Benign | 0.405 | 0.9652 | +0.48 | Neu | |
| c.6529G > A (p.D2177N) | ↑ ATPase [21] | 1.09 × 10−2 | Benign | 0.554 | 0.9683 | −0.02 | Neu | |
| c.6764G > T (p.S2255I) | - | 1.59 × 10−1 | Benign | 0.302 | 0.9619 | +0.13 | Neu | |
| Pathogenic/ Likely pathogenic (P/LP) | c.1804C > T:p(R602W) | ↓ ATPase26 Mislocalization [26,27] | 4.38 × 10−5 | Pathogenic | 0.507 | 0.9034 | +16.56 | Del |
| c.1819G > C (p.G607R) | - | 2.83 × 10−5 | Pathogenic | 0.588 | 0.9447 | +67.4 | Del | |
| c.1957C > T (p.R653C) | ↓ Retinal-stim. ATPase [25,43] | 1.61 × 10−5 | Pathogenic | 0.953 | 0.8201 | +0.8 | Del | |
| c.2894A > G:p(N965S) | ↓ Expression, ↓ ATPase [20,26] | 1.35 × 10−4 | Pathogenic | 0.979 | 0.8819 | +1.1 | Del | |
| c.3352C > T (p.H1118Y) | - | 1.0 × 10−5 | Pathogenic | 0.487 | 0.9513 | +0.15 | Del | |
| c.4462T > C (p.C1488R) | ↓ ATPase20, ↓ ATR binding [28] | 8.20 × 10−6 | Pathogenic | 0.529 | 0.9655 | +21.16 | Del | |
| c.4469G > A (p.C1490Y) | Mislocalization, ↓ ATPase [26] | 5.91 × 10−5 | Pathogenic | 0.345 | 0.9530 | +41.76 | Del | |
| c.5936C > T (p.T1979I) | - | - | Pathogenic | 0.630 | 0.9698 | +2.74 | Del | |
| c.6299G > A (p.G2100E) | - | - | Pathogenic | 0.450 | 0.9139 | +2.85 | Del | |
| c.6316C > T (p.R2106C) | - | 1.31 × 10−4 | Pathogenic | 0.945 | 0.7278 | +3.24 | Del | |
| VUS | c.58A > G (p.R20G) | - | - | Pathogenic | 0.410 | 0.9429 | +2.23 | Del |
| c.294C > G (p.N98K) | - | 1.10 × 10−4 | Benign | 0.503 | 0.9547 | +0.49 | Del | |
| c.1808A > T (p.Y603F) | - | - | Pathogenic b | 0.520 | 0.9567 | −0.75 | Del | |
| c.2252T > C (p.L751P) | - | - | Pathogenic c | 1.019 | 0.7705 | +9.49 | Del | |
| c.2911A > C (p.T971P) | - | - | Pathogenic | 0.559 | 0.9477 | +0.68 | Del | |
| c.3631G > A (p.V1211I) | - | - | Benign | 0.379 | 0.9606 | −3.27 | Neu | |
| c.4672G > A (p.G1558R) | - | - | Pathogenic | 0.625 | 0.9339 | +74.05 | Del | |
| c.5584G > A (p.G1862S) | - | - | Pathogenic ▪ | 0.628 | 0.9181 | N/A | pLoF | |
| c.6320G > A:p(R2107H) | ↓ ATPase26 | 2.03 × 10−3 | Pathogenic | 0.789 | 0.9615 | +1.56 | Del | |
| c.6494A > G (p.Y2165C) | - | 6.57 × 10−6 | Pathogenic | 0.677 | 0.9491 | +1.38 | Del |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cevik, S.; Biswas, S.B.; Biswas-Fiss, E.E. Structural and Pathogenic Impacts of ABCA4 Variants in Retinal Degenerations—An In-Silico Study. Int. J. Mol. Sci. 2023, 24, 7280. https://doi.org/10.3390/ijms24087280
Cevik S, Biswas SB, Biswas-Fiss EE. Structural and Pathogenic Impacts of ABCA4 Variants in Retinal Degenerations—An In-Silico Study. International Journal of Molecular Sciences. 2023; 24(8):7280. https://doi.org/10.3390/ijms24087280
Chicago/Turabian StyleCevik, Senem, Subhasis B. Biswas, and Esther E. Biswas-Fiss. 2023. "Structural and Pathogenic Impacts of ABCA4 Variants in Retinal Degenerations—An In-Silico Study" International Journal of Molecular Sciences 24, no. 8: 7280. https://doi.org/10.3390/ijms24087280