
Synthesis of Novel 2-(Cyclopentylamino)thiazol-4(5H)-one Derivatives with Potential Anticancer, Antioxidant, and 11β-HSD Inhibitory Activities
 , , , and
, , , and 
Abstract
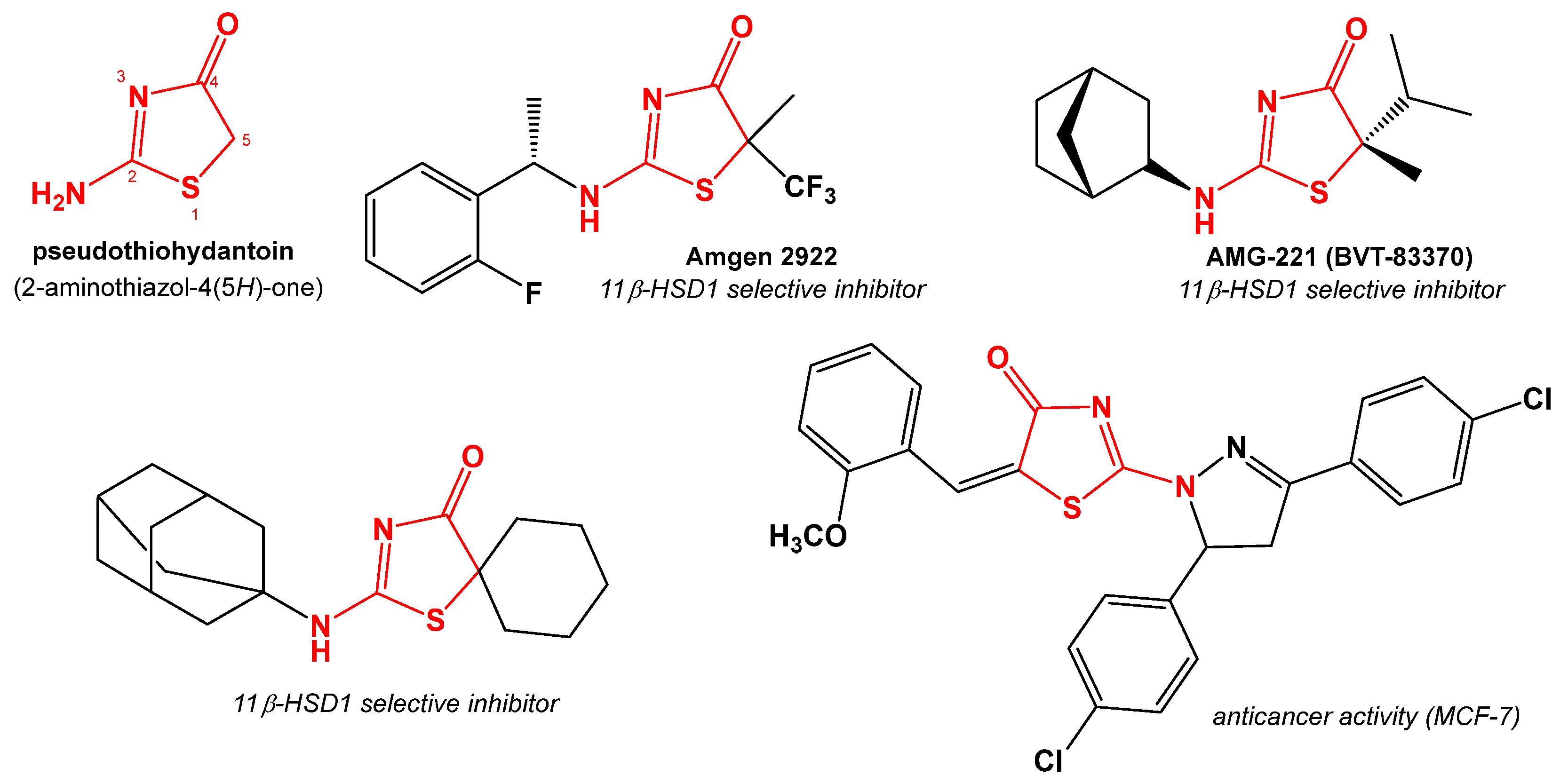
:1. Introduction
2. Results and Discussion
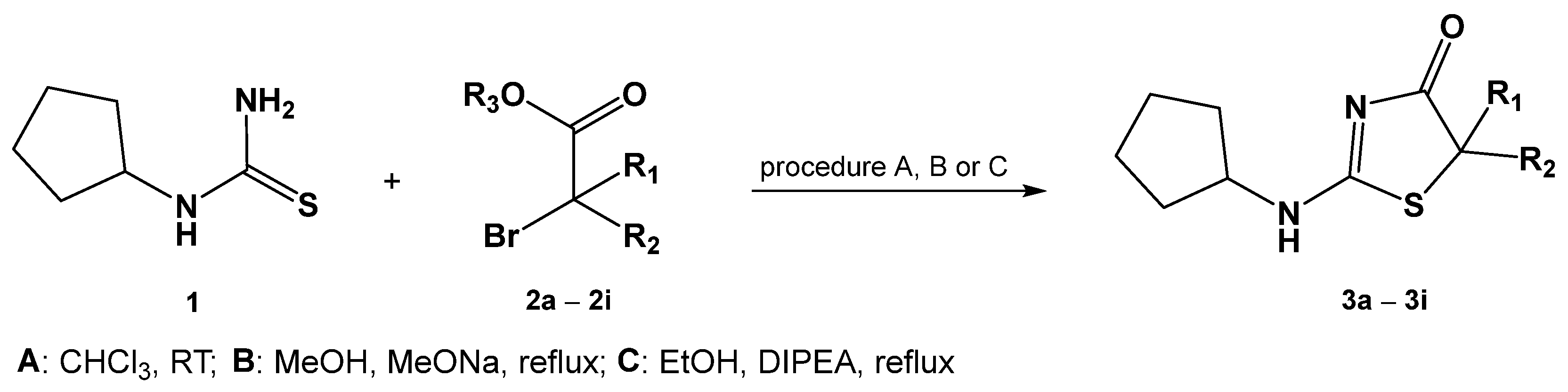
2.1. Chemistry
2.2. Inhibitory Activity towards 11β-HSD
2.2.1. In Vitro Studies
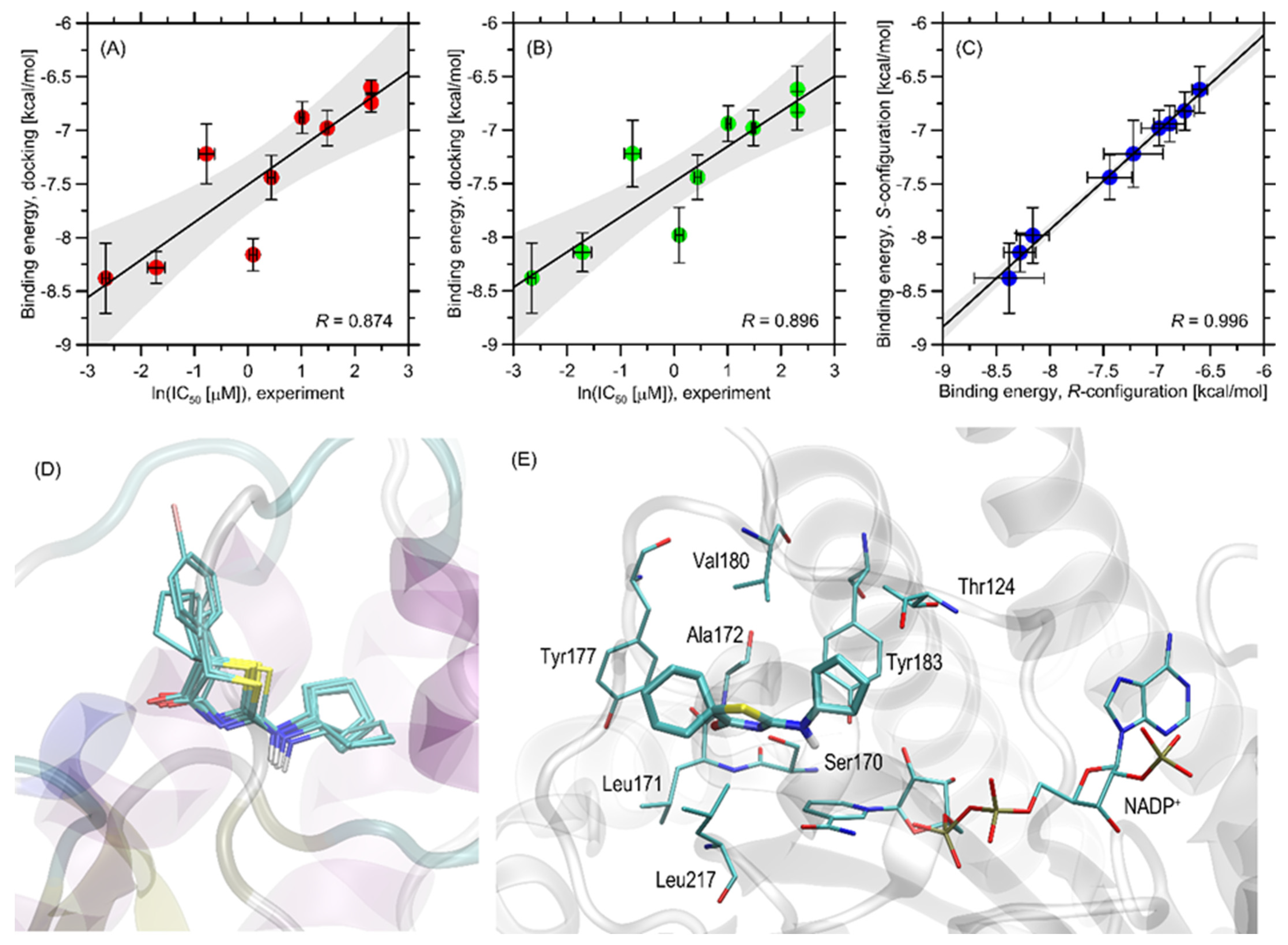
2.2.2. Molecular Docking
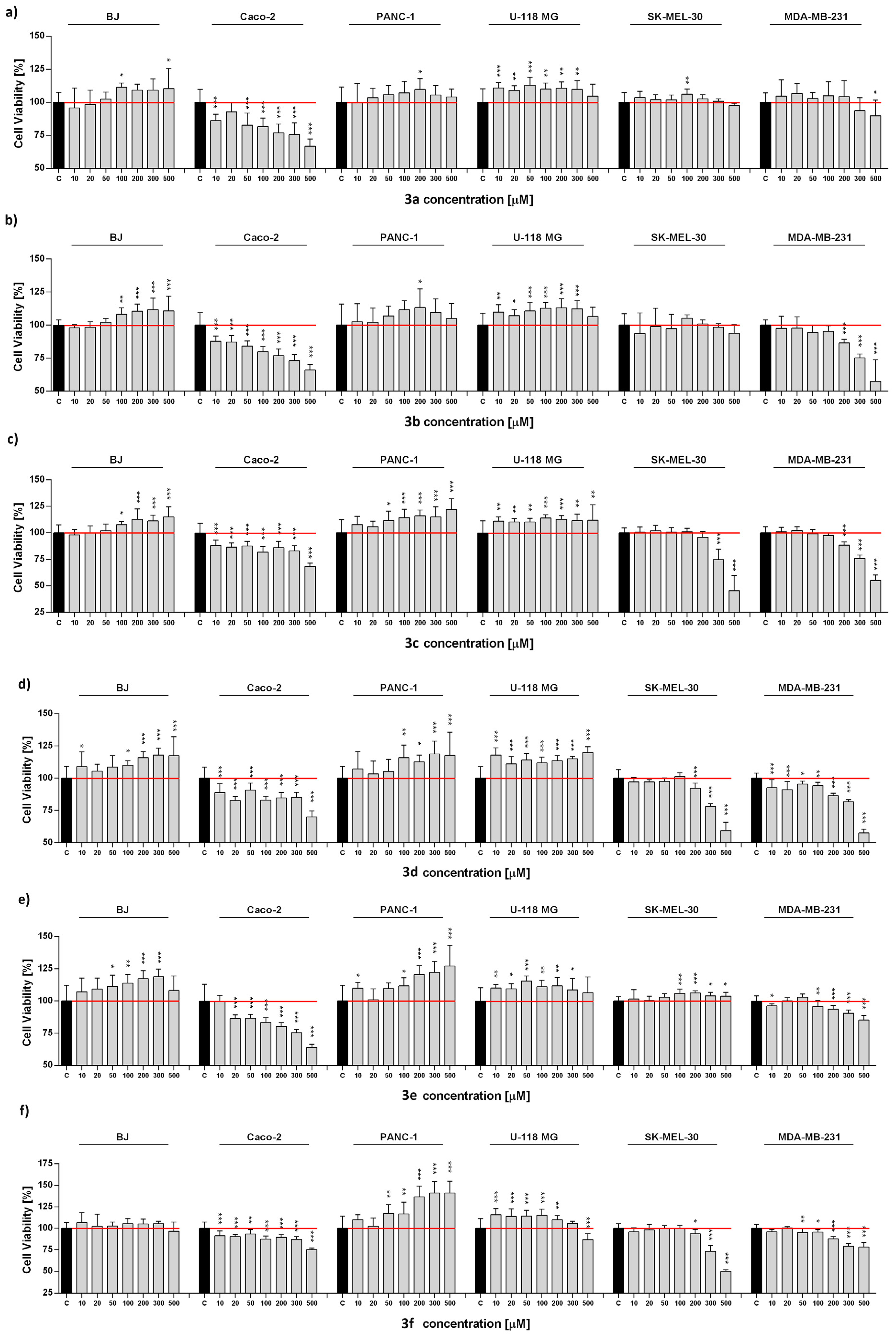
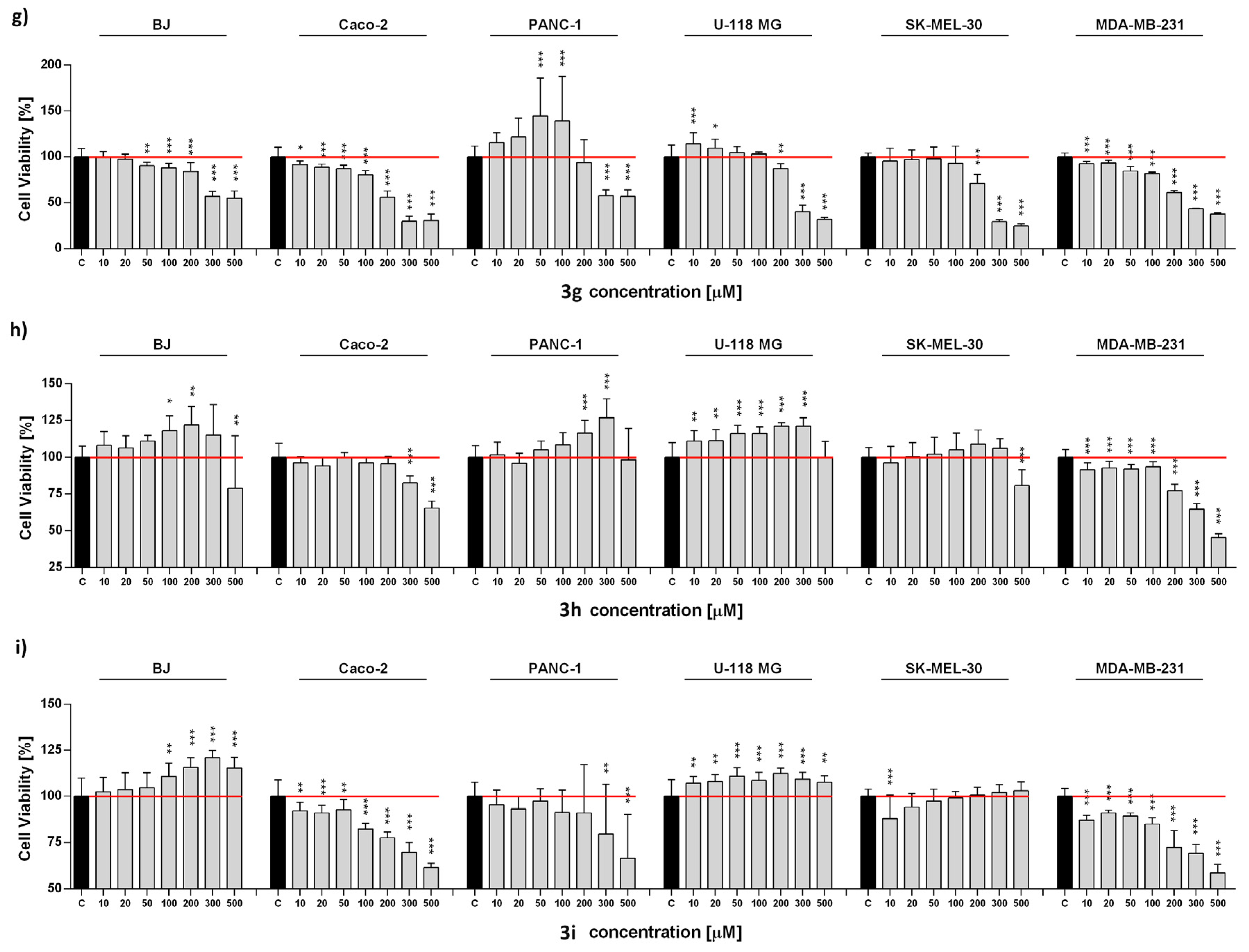
2.3. Anticancer Activity
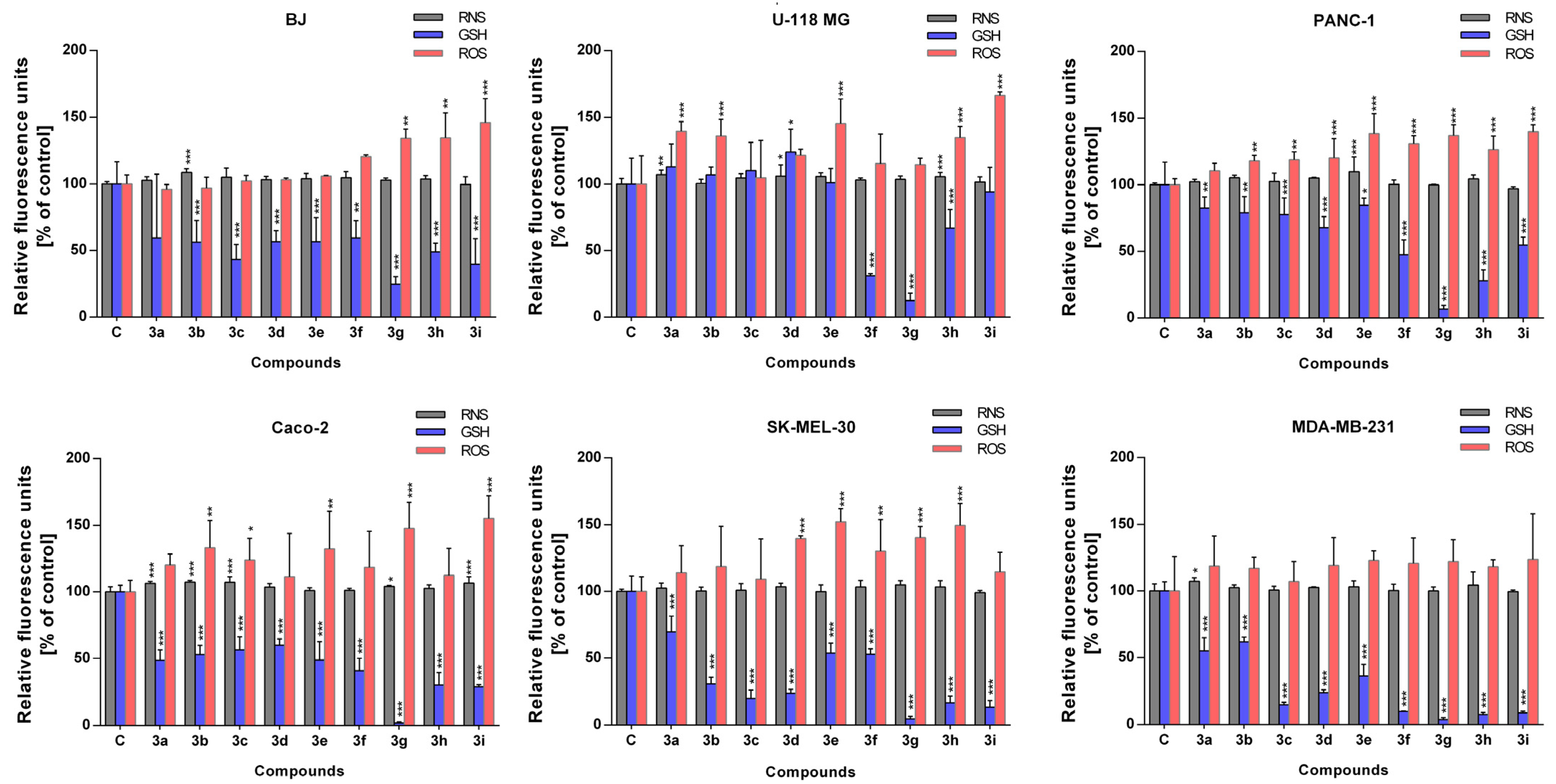
2.4. Antioxidant Activity
3. Materials and Methods
3.1. General Information
3.2. Reagents and Solvents
3.3. General Procedures of Synthesis
3.3.1. Procedure A (Synthesis of Compounds 3a-3c)
3.3.2. Procedure B (Synthesis of Compounds 3d-3e)
3.3.3. Procedure C (Synthesis of Compounds 3f-3i)
3.4. Inhibition of 11β-HSD Assays
3.4.1. 11β-HSD1
3.4.2. 11β-HSD2
3.4.3. Determination of IC50
3.5. Molecular Docking
3.6. Cell Culture
3.7. Metabolic Activity
3.8. Antioxidant Activity
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaikwad, P.L.; Gandhi, P.S.; Jagdale, D.M.; Kadam, V.J. Synthesis, Characterization and In Vitro Antimicrobial Evaluation of Novel Pyrazolothiazol-4(5H)-one Derivatives. Indian J. Pharm. Sci. 2013, 75, 496–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaria, P.N.; Makawana, J.A.; Satasia, S.P.; Raval, D.K.; Zhu, H. Design, synthesis and molecular docking of novel bipyrazolyl thiazolone scaffold as a new class of antibacterial agents. Med. Chem. Commun. 2014, 5, 1555–1562. [Google Scholar] [CrossRef]
- Pansare, D.N.; Shelke, R.N.; Khade, M.C.; Jadhav, V.N.; Pawar, C.D.; Jadhav, R.A.; Bembalkar, S.R. New thiazolone derivatives: Design, synthesis, anticancer and antimicrobial activity. Eur. Chem. Bull. 2019, 8, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, K.; Sharma, M.; Trivedi, P.; Chaturvedi, V.; Chauhan, P.M. New class of methyl tetrazole based hybrid of (Z)-5-benzylidene-2-(piperazin-1-yl)thiazol-4(H)-one as potent antitubercular agents. Bioorg. Med. Chem. Lett. 2014, 24, 4166–4170. [Google Scholar] [CrossRef] [PubMed]
- Nemr, M.T.M.; AboulMagd, A.M.; Hassan, H.M.; Hamed, A.A.; Hamed, M.I.A.; Elsaadi, M.T. Design, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agents via carbonic anhydrase IX inhibition. RSC Adv. 2021, 11, 26241–26257. [Google Scholar] [CrossRef]
- Al-Ansary, G.H.; Ismail, M.A.H.; Abou El Ella, D.A.; Eid, S.; Abouzid, K.A.M. Molecular design and synthesis of HCV inhibitors based on thiazolone scaffold. Eur. J. Med. Chem. 2013, 68, 12–32. [Google Scholar] [CrossRef]
- Yan, S.; Appleby, T.; Larson, G.; Wu, J.Z.; Hamatake, R.; Hong, Z.; Yao, N. Structure-based design of a novel thiazolone scaffold as HCV NS5B polymerase allosteric inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5888–5891. [Google Scholar] [CrossRef]
- Kupczyk, D.; Studzińska, R.; Kołodziejska, R.; Baumgart, S.; Modrzejewska, M.; Woźniak, A. 11β-Hydroxysteroid Dehydrogenase Type 1 as a Potential Treatment Target in Cardiovascular Diseases. J. Clin. Med. 2022, 11, 6190. [Google Scholar] [CrossRef]
- Kupczyk, D.; Bilski, R.; Kozakiewicz, M.; Studzińska, R.; Kędziora-Kornatowska, K.; Kosmalski, T.; Pedrycz-Wieczorska, A.; Głowacka, M. 11β-HSD as a New Target in Pharmacotherapy of Metabolic Diseases. Int. J. Mol. Sci. 2022, 23, 8984. [Google Scholar] [CrossRef]
- El-Sattar, N.E.A.A.; Badawy, E.H.K.; AbdEl-Hady, W.H.; Abo-Alkasem, M.I.; Mandour, A.A.; Ismail, N.S.M. Design and Synthesis of New CDK2 Inhibitors Containing Thiazolone and Thiazolthione Scafold with Apoptotic Activity. Chem. Pharm. Bull. 2021, 69, 106–117. [Google Scholar] [CrossRef]
- Khalil, N.A.; Ahmed, E.M.; El-Nassan, H.B. Synthesis, characterization, and biological evaluation of certain 1,3-thiazolone derivatives bearing pyrazoline moiety as potential anti-breast cancer agents. Med. Chem. Res. 2013, 22, 1021–1027. [Google Scholar] [CrossRef]
- Subtel’na, I.; Atamanyuk, D.; Szymańska, E.; Kieć-Kononowicz, K.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Synthesis of 5-arylidene-2-amino-4-azolones and evaluation of their anticancer activity. Bioorg. Med. Chem. 2010, 18, 5090–5102. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, L.; Le, N.T.; Zhao, C.; Sidduri, A.; Lou, J.P.; Michoud, C.; Portland, L.; Jackson, N.; Liu, J.J.; et al. Synthesis and activity of quinolinyl-methylene-thiazolinones as potent and selective cyclin-dependent kinase 1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 2134–2138. [Google Scholar] [CrossRef] [PubMed]
- Khathi, S.P.; Chandrasekaran, B.; Karunanidhi, S.; Tham, C.L.; Kozielski, F.; Sayyad, N.; Karpoormath, R. Design and synthesis of novel thiadiazole-thiazolone hybrids as potential inhibitors of the human mitotic kinesin Eg5. Bioorg. Med. Chem. Lett. 2018, 28, 2930–2938. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, R.; Kołodziejska, R.; Kupczyk, D.; Plaziński, W.; Kosmalski, T. A novel derivatives of thiazol-4(5H)-one and their activity in the inhibition of 11β-hydroxysteroid dehydrogenase type 1. Bioorganic Chem. 2018, 79, 115–121. [Google Scholar] [CrossRef]
- Studzińska, R.; Kołodziejska, R.; Płaziński, W.; Kupczyk, D.; Kosmalski, T.; Jasieniecka, K.; Modzelewska-Banachiewicz, B. Synthesis of the N-methyl Derivatives of 2-Aminothiazol-4(5H)-one and Their Interactions with 11βHSD1-Molecular Modeling and in Vitro Studies. Chem. Biodivers. 2019, 16, e1900065. [Google Scholar] [CrossRef]
- Kupczyk, D.; Studzińska, R.; Bilski, R.; Baumgart, S.; Kołodziejska, R.; Woźniak, A. Synthesis of novel 2-(isopropylamino)thia-zol-4(5H)-one derivatives and their inhibitory activity of 11β-HSD1 and 11β-HSD2 in aspect of carcinogenesis prevention. Molecules 2020, 25, 4233. [Google Scholar] [CrossRef]
- Kupczyk, D.; Studzińska, R.; Baumgart, S.; Bilski, R.; Kosmalski, T.; Kołodziejska, R.; Woźniak, A. A novel N-tert-butyl derivatives of pseudothiohydantoin as potential target in anti-cancer therapy. Molecules 2021, 26, 2612. [Google Scholar] [CrossRef]
- Studzińska, R.; Kupczyk, D.; Płaziński, W.; Baumgart, S.; Bilski, R.; Paprocka, R.; Kołodziejska, R. Novel 2-(adamantan-1-yloamino)thiazol-4(5H)-one derivatives and their inhibitory activity towards 11β-HSD1—Synthesis molecular docking and in vitro studies. Int. J. Mol. Sci. 2021, 22, 8609. [Google Scholar] [CrossRef]
- Johansson, L.; Fotsch, C.; Bartberger, D.M.; Castro, V.M.; Chen, M.; Emery, M.; Gustafsson, S.; Hale, C.; Hickman, D.; Homan, E.; et al. 2-Amino-1,3-thiazol-4(5H)-ones as potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitors: Enzyme-ligand Co-crystal structure and demonstration of pharmacodynamic effects in C57Bl/6 mice. J. Med. Chem. 2008, 51, 2933–2943. [Google Scholar] [CrossRef]
- Zhunina, O.A.; Yabbarov, N.G.; Grechko, A.V.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Vascular Disease, Tumorigenesis, and Diabetes. Front. Mol. Biosci. 2021, 8, 671908. [Google Scholar] [CrossRef] [PubMed]
- Aouacheria, A.; Baghdiguian, S.; Lamb, H.M.; Huska, J.D.; Pineda, F.J.; Hardwick, J.M. Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem. Int. 2017, 109, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteras, N.; Abramov, A.Y. Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic. Biol. Med. 2022, 189, 136–153. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, CB 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Solek, P.; Mytych, J.; Lannik, E.; Majchrowicz, L.; Koszla, O.; Koziorowska, A.; Koziorowski, M. Cancer on-target: Selective enhancement of 3-bromopyruvate action by an electromagnetic field in vitro. Free Radic. Biol. Med. 2022, 180, 153–164. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.J.; Lee, H.; Hong, S.H.; Park, C.; Park, S.H.; Kim, G.Y.; Kim, S.; Kim, H.S.; Hwang, H.J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Kupczyk, D.; Studzińska, R.; Bilski, R.; Woźniak, A. Application of ELISA technique and human microsomes in the search for 11β-hydroxysteroid dehydrogenase inhibitors. Biomed. Res Int. 2019, 2019, 5747436. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Guo, Z.; Xia, X.; Liu, Y.; Huang, C.; Jiang, L.; Wang, X.; Liu, J.; Huang, H. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J. Exp. Clin. Cancer Res. 2019, 38, 157. [Google Scholar] [CrossRef]
- Solek, P.; Koszla, O.; Mytych, J.; Badura, J.; Chelminiak, Z.; Cuprys, M.; Fraczek, J.; Tabecka-Lonczynska, A.; Koziorowski, M. Neuronal life or death linked to depression treatment: The interplay between drugs and their stress-related outcomes relate to single or combined drug therapies. Apoptosis Int. J. Program. Cell Death 2019, 24, 773–784. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | R1 | R2 | Procedure | Isolated Yield [%] | M.p. (°C) |
|---|---|---|---|---|---|
| 3a | H | CH3 | A | 5.39 * | 127.1–128.9 |
| 3b | H | C2H5 | A | 69.29 * | 132.1–134.0 |
| 3c | H | C3H7 | A | 81.63 * | 112.4–114.0 |
| 3d | H | CH(CH3)2 | B | 19.88 | 135.0–138.0 |
| 3e | CH3 | CH3 | B | 9.42 | 179.0–181.0 |
| 3f | H | C6H5 | C | 71.06 | 183.0–184.0 |
| 3g | H | C6H4p-Br | C | 85.48 | 220 (dec.) |
| 3h | -(CH2)5- | C | 2.47 | 207.7–209.1 | |
| 3i | -(CH2)3- | C | 27.86 | 202.4–203.1 | |
| No. | R1 | R2 | % of 11β-HSD1 Inhibition 10 μM | IC50 11β-HSD1 [µM] | % of 11β-HSD2 Inhibition 10 μM |
|---|---|---|---|---|---|
| 3a | H | CH3 | 10.94 ± 4.08 | >10 | 40.27 ± 0.65 |
| 3b | H | C2H5 | 21.33 ± 3.29 | >10 | 39.70 ± 0.07 |
| 3c | H | C3H7 | 56.58 ± 6.11 | 2.75 ± 0.12 | 37.70 ± 6.07 |
| 3d | H | CH(CH3)2 | 82.93 ± 2.62 | 0.46 ± 0.07 | 38.57 ± 3.69 |
| 3e | CH3 | CH3 | 53.17 ± 5.22 | 4.40 ± 0.15 | 36.61 ± 1.50 |
| 3f | H | C6H5 | 64.29 ± 5.07 | 1.10 ± 0.08 | 36.55 ± 3.07 |
| 3g | H | C6H4p-Br | 86.96 ± 2.17 | 0.18 ± 0.03 | 38.60 ± 1.39 |
| 3h | -(CH2)5- | 90.49 ± 1.31 | 0.07 ± 0.005 | 42.82 ± 0.96 | |
| 3i | -(CH2)3- | 71.25 ± 4.77 | 1.55 ± 0.10 | 46.33 ± 1.22 | |
| Control | 90.42 ± 1.86 a | 0.08 ± 0.006 a | 55.22 ± 0.13 a 46.82 ± 3.75 b | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumgart, S.; Kupczyk, D.; Archała, A.; Koszła, O.; Sołek, P.; Płaziński, W.; Płazińska, A.; Studzińska, R. Synthesis of Novel 2-(Cyclopentylamino)thiazol-4(5H)-one Derivatives with Potential Anticancer, Antioxidant, and 11β-HSD Inhibitory Activities. Int. J. Mol. Sci. 2023, 24, 7252. https://doi.org/10.3390/ijms24087252
Baumgart S, Kupczyk D, Archała A, Koszła O, Sołek P, Płaziński W, Płazińska A, Studzińska R. Synthesis of Novel 2-(Cyclopentylamino)thiazol-4(5H)-one Derivatives with Potential Anticancer, Antioxidant, and 11β-HSD Inhibitory Activities. International Journal of Molecular Sciences. 2023; 24(8):7252. https://doi.org/10.3390/ijms24087252
Chicago/Turabian StyleBaumgart, Szymon, Daria Kupczyk, Aneta Archała, Oliwia Koszła, Przemysław Sołek, Wojciech Płaziński, Anita Płazińska, and Renata Studzińska. 2023. "Synthesis of Novel 2-(Cyclopentylamino)thiazol-4(5H)-one Derivatives with Potential Anticancer, Antioxidant, and 11β-HSD Inhibitory Activities" International Journal of Molecular Sciences 24, no. 8: 7252. https://doi.org/10.3390/ijms24087252