

Regulative Roles of Metabolic Plasticity Caused by Mitochondrial Oxidative Phosphorylation and Glycolysis on the Initiation and Progression of Tumorigenesis

Abstract
:1. Introduction
2. Metabolic Plasticity in the Initiation of Tumourigenesis
2.1. The Importance of Glycolysis and OXPHOS in the Transformation of Stem Cells into Tumour Stem Cells
2.2. Implications and Significance of Research on Metabolic Plasticity of Tumour Stem Cells
3. Molecules Affecting Tumour Glycolysis and OXPHOS Transformation and Significance
3.1. Effect of Glycolytic Rate-Limiting Enzyme on Metabolic Plasticity
3.1.1. Hexokinase
3.1.2. Phosphofructokinase
3.1.3. Pyruvate Kinase
3.2. Effects of Key Enzymes of Mitochondrial Oxidative Metabolism on Metabolic Plasticity
3.2.1. Key Enzymes of the TCA Cycle
Citrate Synthase (CS)
Isocitrate Dehydrogenase (IDH)
α-Ketoglutarate Dehydrogenase Complex(α-KGDC)
3.3. Other Molecules Regulating Metabolism Plasticity Processes
3.4. Summary
4. The Role of Metabolic Plasticity Regulation in the Tumour Microenvironment
4.1. Effects of Metabolic Plasticity Regulation on the Immune Microenvironment
4.2. Effects of Metabolic Plasticity Regulation on Angiogenesis
4.3. Effects of Metabolic Plasticity Regulation on Tumour Migration and Invasion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATPs | Adenosine triphosphates |
| OXPHOS | Oxidative phosphorylation |
| TME | Tumour microenvironment |
| SIRT6 | NAD-dependent protein deacylase sirtuin-6 |
| GLUT3 | Glucose transporter type 3 |
| EGFR | Epidermal growth factor receptor |
| MAPK | MAP kinase-activated protein kinase |
| LT-HSCs | Long-term repopulating haematopoietic stem cells |
| c-Myc | Myc proto-oncogene protein |
| ESC | Epithelial stem cells |
| KRAS | Kirsten rat sarcoma viral oncogene |
| PFK1 | Phosphofructokinase-1 |
| GLUT1 | Glucose transporter type 1 |
| Lgr5 | Leucine rich repeat containing G protein-coupled receptor 5 |
| TFAM | Transcription factor A mitochondrial |
| PARP | Poly [ADP-ribose] polymerase |
| TCA | Tricarboxylic acid |
| PDH | Pyruvate dehydrogenase |
| Nrf-2 | Nuclear factor erythroid 2-related factor 2 |
| TNBC | Triple-negative breast cancer |
| PK | Pyruvate kinase |
| FSH | Follicle-stimulating hormone |
| GBMs | Glioblastoma |
| PDAC | Pancreatic ductal adenocarcinoma |
| hrDNase1 | Human recombinant DNase1 |
| HSP90 | Teat shock protein 90 |
| H2Bub1 | Monoubiquitinated histone H2B |
| VEGF | Vascular endothelial growth factor |
| CAFs | Tumour associated fibroblasts |
| CS | Citrate synthase |
| α-KGDC | α-ketoglutarate dehydrogenase complex |
| CPT | Carnitine palmitoyl transfer I |
| CNS | Central nervous system |
| R-2HG | (R) enantiomer of 2-hydroxyglutarate |
| AML | Acute myeloid leukaemia |
| DMKG | Dimethyl-α-ketoglutarate |
| FBP | Fructose-1, 6-bisphosphatase |
| FH | Fumarate hydratase |
| CoQ10 | Coenzyme Q-binding protein 10 |
| PINK1 | PTEN induced kinase 1 |
| Keap1 | Kelch like ECH associated protein 1 |
| NF1 | Neurofibromin 1 |
| TIGAR | TP53 induced glycolysis regulaTory phosphatase |
| GLS2 | Glutaminase 2 |
| PPARGC-1 | Peroxisome proliferative activated Receptor, gamma, coactivator 1 |
| WTP53 | Wild-type P53 |
| HCC | Hepatocarcinoma |
| Bbc3 | BCL2 binding component 3 |
| PGM | Phosphoglucomutase |
| S1P | Sphingosine 1-phosphate |
| TCR | T cell receptor |
| PTEN | Phosphatase and tensin homolog |
| mTOR | Mechanistic target of rapamycin kinase |
| CIC | Cancer immunity cycle |
| MCT | Monocarboxylate transporter |
| NK | Natural killer |
| IFN-γ | Interferon gamma |
| PGK1 | Phosphoglycerate kinase |
| ER | Endoplasmic reticulum |
| PHD2 | Prolyl hydroxylase-domain 2 |
| ECAR | Extracellular acidification rate |
| PDGF | Platelet-derived growth factor |
| TWIST | twist-related protein |
| Yap | Yes1 associated transcriptional regulator |
| DLK2 | Delta like non-canonical Notch ligand 2 |
| COA3 | Cox assembly factor 3 |
| MMP9 | Matrix metallopeptidase 9 |
| Akt | RAC-beta serine/threonine-protein kinase |
| EMT | Epithelial-mesenchymal transition |
| TICs | Tumour-initiating cells |
| MEIS1 | Homeobox protein Meis1 |
| HSC | Haematopoietic stem cells |
| HIF-1α | Hypoxia inducible factor-1α |
| PDK | Pyruvate dehydrogenase kinase |
| GSCs | Glioma stem cells |
| NF-κB | Nuclear factor NF-kappa-B |
| ISC | Intestinal stem cells |
| HK | Hexokinase |
| LDHA | Lactate dehydrogenase A |
| CBC | Crypt base columnar |
| CRC | Colon epithelial cell carcinoma |
| ROS | Reactive oxygen species |
| LDH | Lactate dehydrogenase |
| PC | Pyruvate carboxylase |
| BCL-2 | B-cell lymphoma-2 |
| AMPK | 5′-AMP-activated protein kinase |
| PFK | Phosphofructokinase |
| VDAC | Voltage-dependent anion channel |
| NSTCs | Non-stem tumour cells |
| PFKP | Phosphofructokinase, platelet |
| NSCLC | Non-small cell lung cancer |
| TRAP1 | TNF receptor associated protein 1 |
| PKM2 | M2 isoform of pyruvate kinase |
| STAT3 | Signal transducer and activator of transcription 3 |
| IGF-IR | Insulin like growth factor 1 receptor |
| COXI | Complex I |
| IDH | Isocitrate dehydrogenase |
| SDH | Successful dehydrogenase |
| Mcl-1 | Myeloid cell leukaemia1 |
| α-KG | α-ketoglutarate |
| LDHB | Lactate dehydrogenase B |
| OCR | Oxygen consumption rate |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NRFs | Nuclear respiratory factors |
| PDSS2 | Decaprenyl diphosphate synthase subunit 2 |
| PEP | Phosphoenolpyruvate |
| TP53 | Tumour protein p53 |
| STK11 | Serine/threonine kinase 11 |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| SCO2 | Synthesis of cytochrome c oxidase 2 |
| G-6-PD | Glucose-6-phosphate dehydrogenase |
| SREBP | Sterol regulatory element binding protein |
| MuTP53 | Mutation in TP53 |
| MPC | Mitochondrial pyruvate carrier |
| AIF | Apoptosis-induced factor |
| CAAs | Cancer-associated antigens |
| IL-7 | Interleukin-7 |
| AGK | Acylglycerol kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| DCs | Dendritic cells |
| PD-L1 | Programmed cell death 1 ligand 1 |
| Treg | Regulatory T |
| ARG1 | Arginase 1 |
| PDPK1 | Phosphoinositide dependent protein kinase 1 |
| TGF-β | Transforming growth factor beta |
| bFGF | Fibroblast growth factor 2 |
| PFKFB3 | Protein 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 |
| LA | Lactic acid |
| SNAI | Snail family transcriptional repressor |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
| TAZ | Tafazzin |
| EGF | Epithelial growth factor |
| SLC1A5 | Sodium-dependent neutral amino acid transporter type 2 |
| CD28 | Leukocyte surface differentiation antigen 28 |
References
- Bausewein, T.; Nussberger, S.; Kuhlbrandt, W. Cryo-EM structure of Neurospora crassa respiratory complex IV. IUCr J. 2019, 6 Pt 4, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131 Pt 7, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kim, J. Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 2019, 8, 275. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Pouysségur, J.; Marchiq, I.; Parks, S.K.; Durivault, J.; Ždralević, M.; Vucetic, M. Warburg effect’ controls tumor growth, bacterial, viral infections and immunity–Genetic deconstruction and therapeutic perspectives. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022. [Google Scholar]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef]
- Yang, J.; Liu, D.J.; Zheng, J.H.; He, R.Z.; Xu, D.P.; Yang, M.W.; Yao, H.F.; Fu, X.L.; Yang, J.Y.; Huo, Y.M.; et al. IRAK2-NF-kappaB signaling promotes glycolysis-dependent tumor growth in pancreatic cancer. Cell Oncol. 2022, 45, 367–379. [Google Scholar] [CrossRef]
- Reinfeld, B.I.; Rathmell, W.K.; Kim, T.K.; Rathmell, J.C. The therapeutic implications of immunosuppressive tumor aerobic glycolysis. Cell Mol. Immunol. 2022, 19, 46–58. [Google Scholar] [CrossRef]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Yoon, J.H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.J.; Liu, W.; Welch, D.R. The KISS1 metastasis suppressor appears to reverse the Warburg effect by shifting from glycolysis to mitochondrial beta-oxidation. J. Mol. Med. 2017, 95, 951–963. [Google Scholar] [CrossRef]
- Yuen, C.A.; Asuthkar, S.; Guda, M.R.; Tsung, A.J.; Velpula, K.K. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: A new target gleaned from an old concept. CNS Oncol. 2016, 5, 101–108. [Google Scholar] [CrossRef]
- Xu, J.; Richard, S. Cellular pathways influenced by protein arginine methylation: Implications for cancer. Mol. Cell 2021, 81, 4357–4368. [Google Scholar] [CrossRef]
- Chembazhi, U.V.; Bangru, S.; Hernaez, M.; Kalsotra, A. Cellular plasticity balances the metabolic and proliferation dynamics of a regenerating liver. Genome Res. 2021, 31, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.; Stanger, B. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenring, J.R.; Mills, J.C. Cellular Plasticity, Reprogramming, and Regeneration: Metaplasia in the Stomach and Beyond. Gastroenterology 2022, 162, 415–430. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Liu, C.; Qiang, J.; Deng, Q.; Xia, J.; Deng, L.; Zhou, L.; Wang, D.; He, X.; Liu, Y.; Zhao, B.; et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res. 2021, 81, 5919–5934. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Tateishi, K.; Kato, H.; Fujiwara, H.; Yamamoto, K.; Kudo, Y.; Nakagawa, H.; Tanaka, Y.; Ijichi, H.; Ikenoue, T.; et al. Inhibition of histone methyltransferase G9a attenuates liver cancer initiation by sensitizing DNA-damaged hepatocytes to p53-induced apoptosis. Cell Death Dis. 2021, 12, 99. [Google Scholar] [CrossRef]
- Inamura, K. Clinicopathological Characteristics and Mutations Driving Development of Early Lung Adenocarcinoma: Tumor Initiation and Progression. Int. J. Mol. Sci. 2018, 19, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.W.; Ma, C.; Medoro, L.; Chen, L.; Wang, B.; Gupta, R.; Liu, T.; Yang, X.Z.; Chen, T.T.; Wang, R.Z.; et al. LncRNA ANRIL is up-regulated in nasopharyngeal carcinoma and promotes the cancer progression via increasing proliferation, reprograming cell glucose metabolism and inducing side-population stem-like cancer cells. Oncotarget 2016, 7, 61741–61754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander, H.M. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Lyssiotis, C.A.; Cantley, L.C. SIRT6 puts cancer metabolism in the driver’s seat. Cell 2012, 151, 1155–1156. [Google Scholar] [CrossRef] [Green Version]
- Onodera, Y.; Nam, J.M.; Bissell, M.J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 2014, 124, 367–384. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2016, 15, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; Ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, G.; Zheng, L.; Zhou, Y.; Sheng, H.; Wu, L.; Zhang, Q.; Lei, J.; Zhang, J.; Xin, R.; et al. STIM1 is a metabolic checkpoint regulating the invasion and metastasis of hepatocellular carcinoma. Theranostics 2020, 10, 6483–6499. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Konno, Y.; Ihira, K.; Kobayashi, N.; Yue, J.; Watari, H. Long non-coding RNA DLEU2 drives EMT and glycolysis in endometrial cancer through HK2 by competitively binding with miR-455 and by modulating the EZH2/miR-181a pathway. J. Exp. Clin. Cancer Res. 2021, 40, 216. [Google Scholar] [CrossRef]
- Park, H.A.; Brown, S.R.; Kim, Y. Cellular Mechanisms of Circulating Tumor Cells During Breast Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 5040. [Google Scholar] [CrossRef]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Metabolic reprogramming sustains cancer cell survival following extracellular matrix detachment. Redox Biol. 2020, 36, 101643. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todaro, G.J.; Huebner, R.J. NAS symposium: New evidence as the basis for increased efforts in cancer research. Proc. Natl. Acad. Sci. USA 1972, 69, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelek, J.M. Cancer-cell fusion with migratory bone-marrow-derived cells as an explanation for metastasis: New therapeutic paradigms. Future Oncol. 2008, 4, 449–452. [Google Scholar] [CrossRef]
- Knudson, A.J. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Potter, V.R. Phenotypic diversity in experimental hepatomas: The concept of partially blocked ontogeny. The 10th Walter Hubert Lecture. Br. J. Cancer 1978, 38, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, G.B. Relationship between differentiation and carcinogenesis. J. Toxicol. Environ. Health 1977, 2, 1335–1342. [Google Scholar] [CrossRef]
- Zhao, Z.; Zuber, J.; Diaz-Flores, E.; Lintault, L.; Kogan, S.C.; Shannon, K.; Lowe, S.W. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010, 24, 1389–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [Green Version]
- Martin, U. Genome stability of programmed stem cell products. Adv. Drug. Deliv. Rev. 2017, 120, 108–117. [Google Scholar] [CrossRef]
- Yu, X.; Mengsteab, P.Y.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Enhancing the Surface Properties of a Bioengineered Anterior Cruciate Ligament Matrix for Use with Point-of-Care Stem Cell Therapy. Engineering 2021, 7, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Halley-Stott, R.P.; Pasque, V.; Gurdon, J.B. Nuclear reprogramming. Development 2013, 140, 2468–2471. [Google Scholar] [CrossRef] [Green Version]
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef] [Green Version]
- Pawelek, J.M. Tumour cell hybridization and metastasis revisited. Melanoma Res. 2000, 10, 507–514. [Google Scholar] [CrossRef]
- Rizvi, A.Z.; Swain, J.R.; Davies, P.S.; Bailey, A.S.; Decker, A.D.; Willenbring, H.; Grompe, M.; Fleming, W.H.; Wong, M.H. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6321–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aractingi, S.; Kanitakis, J.; Euvrard, S.; Le Danff, C.; Peguillet, I.; Khosrotehrani, K.; Lantz, O.; Carosella, E.D. Skin carcinoma arising from donor cells in a kidney transplant recipient. Cancer Res. 2005, 65, 1755–1760. [Google Scholar] [CrossRef] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimmeck, D.; Hansson, J.; Raffel, S.; Vakhrushev, S.Y.; Trumpp, A.; Krijgsveld, J. Proteomic cornerstones of hematopoietic stem cell differentiation: Distinct signatures of multipotent progenitors and myeloid committed cells. Mol. Cell. Proteomics 2012, 11, 286–302. [Google Scholar] [CrossRef] [Green Version]
- Morfouace, M.; Lalier, L.; Bahut, M.; Bonnamain, V.; Naveilhan, P.; Guette, C.; Oliver, L.; Gueguen, N.; Reynier, P.; Vallette, F.M. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J. Biol. Chem. 2012, 287, 33664–33674. [Google Scholar] [CrossRef] [Green Version]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Viola, G.; Della Puppa, A.; Cavallini, L.; Frasson, C.; Persano, L.; Panchision, D.M.; Basso, G. Hypoxia and succinate antagonize 2-deoxyglucose effects on glioblastoma. Biochem. Pharmacol. 2010, 80, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, H.; Chen, S.; Lu, Z.; Li, B.; Jiang, T.; Xuan, M.; Ye, R.; Liang, H.; Liu, X.; et al. SIRT1 regulated hexokinase-2 promoting glycolysis is involved in hydroquinone-enhanced malignant progression in human lymphoblastoid TK6 cells. Ecotoxicol. Environ. Saf. 2022, 241, 113757. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wei, Y.; Hu, B.; Liao, Y.; Wang, X.; Wan, W.H.; Huang, C.X.; Mahabati, M.; Liu, Z.Y.; Qu, J.R.; et al. c-Myc-driven glycolysis polarizes functional regulatory B cells that trigger pathogenic inflammatory responses. Signal Transduct. Target. Ther. 2022, 7, 105. [Google Scholar] [CrossRef]
- Qu, X.; Sun, J.; Zhang, Y.; Li, J.; Hu, J.; Li, K.; Gao, L.; Shen, L. c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 415–421. [Google Scholar] [CrossRef]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formica, V.; Sera, F.; Cremolini, C.; Riondino, S.; Morelli, C.; Arkenau, H.T.; Roselli, M. KRAS and BRAF Mutations in Stage II and III Colon Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2022, 114, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Judd, J.; Abdel Karim, N.; Khan, H.; Naqash, A.R.; Baca, Y.; Xiu, J.; VanderWalde, A.M.; Mamdani, H.; Raez, L.E.; Nagasaka, M.; et al. Characterization of KRAS Mutation Subtypes in Non-small Cell Lung Cancer. Mol. Cancer Ther. 2021, 20, 2577–2584. [Google Scholar] [CrossRef]
- Sasaki, H.; Shitara, M.; Yokota, K.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. Overexpression of GLUT1 correlates with Kras mutations in lung carcinomas. Mol. Med. Rep. 2012, 5, 599–602. [Google Scholar] [CrossRef]
- Lefort, S.; Tan, S.; Balani, S.; Rafn, B.; Pellacani, D.; Hirst, M.; Sorensen, P.H.; Eaves, C.J. Initiation of human mammary cell tumorigenesis by mutant KRAS requires YAP inactivation. Oncogene 2020, 39, 1957–1968. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Kemper, K.; Prasetyanti, P.R.; De Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 2012, 30, 2378–2386. [Google Scholar] [CrossRef]
- Merlos-Suárez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Céspedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Muñoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnay, F.; Veloso, A.; Steinmann, V.; Köcher, T.; Abdusselamoglu, M.D.; Bajaj, S.; Rivelles, E.; Landskron, L.; Esterbauer, H.; Zinzen, R.P.; et al. Oxidative Metabolism Drives Immortalization of Neural Stem Cells during Tumorigenesis. Cell 2020, 182, 1490–1507.e19. [Google Scholar] [CrossRef] [PubMed]
- Hottiger, M.O. Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annu. Rev. Biochem. 2015, 84, 227–263. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivillibhuthur, M.; Warder, B.N.; Toke, N.H.; Shah, P.P.; Feng, Q.; Gao, N.; Bonder, E.M.; Verzi, M.P. TFAM is required for maturation of the fetal and adult intestinal epithelium. Dev. Biol. 2018, 439, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2020, 32, 107793. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.; Mondragón, L.; Pranjic, B.; Hanada, T.; Stoll, G.; Köcher, T.; Zhang, P.; Jais, A.; Lercher, A.; Bergthaler, A.; et al. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Res. 2019, 29, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastò, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisieux, A.; Pommier, R.M.; Morel, A.P.; Lavial, F. Cellular Pliancy and the Multistep Process of Tumorigenesis. Cancer Cell 2018, 33, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Fan, Y.; Li, D.; Han, B.; Meng, Y.; Chen, F.; Liu, T.; Song, Z.; Han, Y.; Huang, L.; et al. Ferroptosis inducer erastin sensitizes NSCLC cells to celastrol through activation of the ROS-mitochondrial fission-mitophagy axis. Mol. Oncol. 2021, 15, 2084–2105. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Saravanan, K.; Mothana, R.A.; Ramachandran, G.; Rajivgandhi, G.; Manoharan, N. Anti-cancer activity of biosynthesized silver nanoparticles using Avicennia marina against A549 lung cancer cells through ROS/mitochondrial damages. Saudi J. Biol. Sci. 2020, 27, 3018–3024. [Google Scholar] [CrossRef]
- Pacchiana, R.; Mullappilly, N.; Pinto, A.; Bova, S.; Forciniti, S.; Cullia, G.; Dalla Pozza, E.; Bottani, E.; Decimo, I.; Dando, I.; et al. 3-Bromo-Isoxazoline Derivatives Inhibit GAPDH Enzyme in PDAC Cells Triggering Autophagy and Apoptotic Cell Death. Cancers 2022, 14, 3153. [Google Scholar] [CrossRef]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., 3rd; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.; Tang, D.; Paik, J. Oxidative stress sensing and response in neural stem cell fate. Free Radic Biol. Med. 2021, 169, 74–83. [Google Scholar] [CrossRef]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef]
- Saga, I.; Shibao, S.; Okubo, J.; Osuka, S.; Kobayashi, Y.; Yamada, S.; Fujita, S.; Urakami, K.; Kusuhara, M.; Yoshida, K.; et al. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol. 2014, 16, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensard, C.L.; Wisidagama, D.R.; Olson, K.A.; Berg, J.A.; Krah, N.M.; Schell, J.C.; Nowinski, S.M.; Fogarty, S.; Bott, A.J.; Wei, P.; et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab. 2020, 31, 284–300.e7. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, I.; Bonnay, F.; Steinmann, V.; Loedige, I.; Burkard, T.R.; Meister, G.; Knoblich, J.A. The tumor suppressor Brat controls neuronal stem cell lineages by inhibiting Deadpan and Zelda. EMBO Rep. 2018, 19, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Rehman, M.T.; Ismael, M.A.; AlAjmi, M.F.; Alruwaished, G.I.; Alokail, M.S.; Khan, M.R. Bioflavonoid (Hesperidin) Restrains Protein Oxidation and Advanced Glycation End Product Formation by Targeting AGEs and Glycolytic Enzymes. Cell Biochem. Biophys. 2021, 79, 833–844. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587 Pt 23, 5591–5600. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Razmpour, R.; Lutton, E.; Alfonso-Prieto, M.; Ramirez, S.H.; Gamero, A.M. Characterization of cancer-associated IDH2 mutations that differ in tumorigenicity, chemosensitivity and 2-hydroxyglutarate production. Oncotarget 2019, 10, 2675–2692. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Nakagawa, M.; Nakatani, F.; Matsunaga, H.; Seki, T.; Endo, M.; Ogawara, Y.; Machida, Y.; Katsumoto, T.; Yamagata, K.; Hattori, A.; et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene 2019, 38, 6835–6849. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Kim, C.F.; Dirks, P.B. Cancer and stem cell biology: How tightly intertwined? Cell Stem Cell 2008, 3, 147–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Shibao, S.; Minami, N.; Koike, N.; Fukui, N.; Yoshida, K.; Saya, H.; Sampetrean, O. Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol. 2018, 20, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin. Cancer Biol. 2022, 87, 184–195. [Google Scholar] [CrossRef]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, A.; Sandoval-Gonzalez, S.; Takahashi, R.; Krall, A.; Sathe, L.; Wei, L.; Radu, C.; Joly, J.H.; Graham, N.A.; Christofk, H.R.; et al. Increased lactate dehydrogenase activity is dispensable in squamous carcinoma cells of origin. Nat. Commun. 2019, 10, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellers, K.; Fox, M.P.; Bousamra, M., 2nd; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Herschman, H.R. A Tumor Agnostic Therapeutic Strategy for Hexokinase 1-Null/Hexokinase 2-Positive Cancers. Cancer Res. 2019, 79, 5907–5914. [Google Scholar] [CrossRef] [Green Version]
- Fukushi, A.; Kim, H.D.; Chang, Y.C.; Kim, C.H. Revisited Metabolic Control and Reprogramming Cancers by Means of the Warburg Effect in Tumor Cells. Int. J. Mol. Sci. 2022, 23, 10037. [Google Scholar] [CrossRef]
- Zhang, R.; Shen, W.; Du, J.; Gillies, M.C. Selective knockdown of hexokinase 2 in rods leads to age-related photoreceptor degeneration and retinal metabolic remodeling. Cell Death Dis. 2020, 11, 885. [Google Scholar] [CrossRef]
- Petit, L.; Ma, S.; Cipi, J.; Cheng, S.Y.; Zieger, M.; Hay, N.; Punzo, C. Aerobic Glycolysis Is Essential for Normal Rod Function and Controls Secondary Cone Death in Retinitis Pigmentosa. Cell Rep. 2018, 23, 2629–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, L.; Ma, S.; Cipi, J.; Cheng, S.Y.; Zieger, M.; Hay, N.; Punzo, C. Monoamine Oxidase A is a Major Mediator of Mitochondrial Homeostasis and Glycolysis in Gastric Cancer Progression. Cancer Manag. Res. 2020, 12, 8023–8035. [Google Scholar]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): Function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–22470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 2009, 1787, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [Green Version]
- Cesar, M.C.; Wilson, J.E. Further studies on the coupling of mitochondrially bound hexokinase to intramitochondrially compartmented ATP, generated by oxidative phosphorylation. Arch Biochem. Biophys. 1998, 350, 109–117. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef]
- Yam, M.; Engel, A.L.; Wang, Y.; Zhu, S.; Hauer, A.; Zhang, R.; Lohner, D.; Huang, J.; Dinterman, M.; Zhao, C.; et al. Proline mediates metabolic communication between retinal pigment epithelial cells and the retina. J. Biol. Chem. 2019, 294, 10278–10289. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta 2016, 1857, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Q.; Zhang, M.; Cao, C.; Liu, X.; Zhang, M.; Li, G.; Xu, C.; Zhang, X. Enhanced antitumor effects of follicle-stimulating hormone receptor-mediated hexokinase-2 depletion on ovarian cancer mediated by a shift in glucose metabolism. J. Nanobiotechnology 2020, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N.; et al. Author Correction: Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, C.S.S.; Sousa, S.F. Current Status of the Use of Multifunctional Enzymes as Anti-Cancer Drug Targets. Pharmaceutics. 2021, 14, 10. [Google Scholar] [CrossRef]
- Zhou, K.; Yao, Y.L.; He, Z.C.; Chen, C.; Zhang, X.N.; Yang, K.D.; Liu, Y.Q.; Liu, Q.; Fu, W.J.; Chen, Y.P.; et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018, 9, 988. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zou, L.; Lu, G.; Grinchuk, O.; Fang, L.; Ong, D.S.T.; Taneja, R.; Ong, C.N.; Shen, H.M. PFKP alleviates glucose starvation-induced metabolic stress in lung cancer cells via AMPK-ACC2 dependent fatty acid oxidation. Cell Discov. 2022, 8, 52. [Google Scholar] [CrossRef]
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic Regulation of Apoptosis in Cancer. Int. Rev. Cell Mol. Biol. 2016, 327, 43–87. [Google Scholar]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Yang, Y.; Ishak Gabra, M.B.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Li, H.; Milman, N.; Liu, J.; Reid, M.A.; Locasale, J.W.; et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat. Commun. 2019, 10, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddalena, F.; Condelli, V.; Matassa, D.S.; Pacelli, C.; Scrima, R.; Lettini, G.; Li Bergolis, V.; Pietrafesa, M.; Crispo, F.; Piscazzi, A.; et al. TRAP1 enhances Warburg metabolism through modulation of PFK1 expression/activity and favors resistance to EGFR inhibitors in human colorectal carcinomas. Mol. Oncol. 2020, 14, 3030–3047. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyel, P.A. Dnases in health and disease. Dev. Biol. 2017, 429, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, H.; Li, X.; Mo, X.; Zhang, H.; Yang, L.; Deng, Y.; Yan, Y.; Yang, G.; Liu, X.; et al. DNASE1L3 as an indicator of favorable survival in hepatocellular carcinoma patients following resection. Aging 2020, 12, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lv, W.; Li, X.; Zhang, L.; Lin, J. Prognostic genes of hepatocellular carcinoma based on gene coexpression network analysis. J. Cell Biochem. 2019, 120, 11616–11623. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yang, K.; Liu, P.; Ma, D.; Lei, P.; Liu, Q. Deoxyribonuclease 1-like 3 Inhibits Hepatocellular Carcinoma Progression by Inducing Apoptosis and Reprogramming Glucose Metabolism. Int. J. Biol. Sci. 2022, 18, 82–95. [Google Scholar] [CrossRef]
- Rosner, K. DNase1: A new personalized therapy for cancer? Expert Rev. Anticancer Ther. 2011, 11, 981–984. [Google Scholar] [CrossRef]
- Joshi, A.; Dai, L.; Liu, Y.; Lee, J.; Ghahhari, N.M.; Segala, G.; Beebe, K.; Jenkins, L.M.; Lyons, G.C.; Bernasconi, L.; et al. The mitochondrial HSP90 paralog TRAP1 forms an OXPHOS-regulated tetramer and is involved in mitochondrial metabolic homeostasis. BMC Biol. 2020, 18, 10. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, Y.; Wang, Q.; Qu, J.; Wei, X.; Xu, J.; Wang, Y.; Suo, F.; Zhang, Y. High developmental pluripotency-associated 4 expression promotes cell proliferation and glycolysis, and predicts poor prognosis in non-small-cell lung cancer. Mol. Med. Rep. 2019, 20, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.Y.; Cai, F.F.; Zhang, L.; Han, J.; Yang, L.; Tang, F.; Li, Y.B.; Chang, J.F.; Sun, F.; Yang, X.M.; et al. Epigenetic regulation of the Warburg effect by H2B monoubiquitination. Cell Death Differ. 2020, 27, 1660–1676. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Han, J.; Jia, L.; Hu, X.; Chen, L.; Wang, Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell 2019, 10, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, H.; Li, Y.; Tan, M.; Fan, S.; Cao, C.; Meng, F.; Zhu, L.; Zhao, L.; Guan, M.X.; et al. Inhibiting neddylation modification alters mitochondrial morphology and reprograms energy metabolism in cancer cells. JCI Insight 2019, 4, e121582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 98–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Gui, D.Y.; Lewis, C.A.; Vander Heiden, M.G. Allosteric regulation of PKM2 allows cellular adaptation to different physiological states. Sci. Signal 2013, 6, e7. [Google Scholar] [CrossRef]
- Feng, C.; Gao, Y.; Wang, C.; Yu, X.; Zhang, W.; Guan, H.; Shan, Z.; Teng, W. Aberrant overexpression of pyruvate kinase M2 is associated with aggressive tumor features and the BRAF mutation in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1524–E1533. [Google Scholar] [CrossRef] [Green Version]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [Green Version]
- Amin, S.; Yang, P.; Li, Z. Pyruvate kinase M2: A multifarious enzyme in non-canonical localization to promote cancer progression. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, L.Z.; Yin, Y.; He, J.; Li, Q.; Qian, X.; You, Y.; Lu, Z.; Peiper, S.C.; Shu, Y.; et al. Regulatory circuit of PKM2/NF-kappaB/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene 2015, 34, 5482–5493. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, P.; Li, Z. The multifaceted regulation and functions of PKM2 in tumor progression. Biochim. Biophys. Acta 2014, 1846, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, Y.; Qiao, J.; Yang, J.J.; Liu, Z.R. Pyruvate kinase M2 in blood circulation facilitates tumor growth by promoting angiogenesis. J. Biol. Chem. 2014, 289, 25812–25821. [Google Scholar] [CrossRef] [Green Version]
- Azoitei, N.; Becher, A.; Steinestel, K.; Rouhi, A.; Diepold, K.; Genze, F.; Simmet, T.; Seufferlein, T. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol. Cancer 2016, 15, 3. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Taddei, M.L.; Morandi, A.; Comito, G.; Calvani, M.; Bianchini, F.; Richichi, B.; Raugei, G.; Wong, N.; Tang, D.; et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 2015, 6, 24061–24074. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal 2018, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar]
- Curry, J.M.; Tuluc, M.; Whitaker-Menezes, D.; Ames, J.A.; Anantharaman, A.; Butera, A.; Leiby, B.; Cognetti, D.M.; Sotgia, F.; Lisanti, M.P.; et al. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle 2013, 12, 1371–1384. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 4777–47795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Aghamir, S.; Tavangar, S.M. Oncometabolites: A new insight for oncology. Mol. Genet. Genomic. Med. 2019, 7, e873. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Limonta, P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free Radic. Biol. Med. 2021, 176, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.W.; Jeong, Y.J.; Park, S.H.; Oh, H.K.; Kang, S. Reverse Warburg Effect-Related Mitochondrial Activity and (18)F-FDG Uptake in Invasive Ductal Carcinoma. Nucl. Med. Mol. Imaging 2019, 53, 396–405. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic Heterogeneity of Cancer Cells: An Interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef] [Green Version]
- Sajnani, K.; Islam, F.; Smith, R.A.; Gopalan, V.; Lam, A.K. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie 2017, 135, 164–172. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Chesney, J. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Infantino, V. Citrate—New functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef]
- Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. [Google Scholar] [CrossRef]
- Corrêa-Ferreira, M.L.; do Rocio Andrade Pires, A.; Barbosa, I.R.; Echevarria, A.; Pedrassoli, G.H.; Winnischofer, S.M.B.; Noleto, G.R.; Cadena, S.M.S.C. The mesoionic compound MI-D changes energy metabolism and induces apoptosis in T98G glioma cells. Mol. Cell Biochem. 2022, 477, 2033–2045. [Google Scholar] [CrossRef]
- Liang, L.; Zhang, G.; Cheng, C.; Li, H.; Jin, T.; Su, C.; Xiao, Y.; Bradley, J.; Peberdy, M.A.; Ornato, J.P.; et al. High-resolution respirometry for evaluation of mitochondrial function on brain and heart homogenates in a rat model of cardiac arrest and cardiopulmonary resuscitation. Biomed. Pharmacother. 2021, 142, 111935. [Google Scholar] [CrossRef] [PubMed]
- Djafarzadeh, S.; Jakob, S.M. High-resolution Respirometry to Assess Mitochondrial Function in Permeabilized and Intact Cells. J. Vis. Exp. 2017, 120, e54985. [Google Scholar]
- Lu, Y.; Zhang, X.; Zhang, H.; Lan, J.; Huang, G.; Varin, E.; Lincet, H.; Poulain, L.; Icard, P. Citrate induces apoptotic cell death: A promising way to treat gastric carcinoma? Anticancer Res. 2011, 31, 797–805. [Google Scholar] [PubMed]
- Yousefi, S.; Owens, J.W.; Cesario, T.C. Citrate shows specific, dose-dependent lympholytic activity in neoplastic cell lines. Leuk. Lymphoma 2004, 45, 1657–1665. [Google Scholar] [CrossRef]
- Zhang, X.; Varin, E.; Allouche, S.; Lu, Y.; Poulain, L.; Icard, P. Effect of citrate on malignant pleural mesothelioma cells: A synergistic effect with cisplatin. Anticancer Res. 2009, 29, 1249–1254. [Google Scholar]
- Kruspig, B.; Nilchian, A.; Orrenius, S.; Zhivotovsky, B.; Gogvadze, V. Citrate kills tumor cells through activation of apical caspases. Cell Mol. Life Sci. 2012, 69, 4229–4237. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Machida, S.; Kondo, M.; Terasaki, H.; Yokoyama, D.; Kurosaka, D. Enhancement of ON-bipolar cell responses of cone electroretinograms in rabbits with the Pro347Leu rhodopsin mutation. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7610–7617. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.I.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.A.; Zhang, X.D.; Guo, X.Y.; Xian, S.L.; Lu, Y.F. 3-bromopyruvate and sodium citrate target glycolysis, suppress survivin, and induce mitochondrial-mediated apoptosis in gastric cancer cells and inhibit gastric orthotopic transplantation tumor growth. Oncol. Rep. 2016, 35, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Halabe, B.A. Hypothesis proved...citric acid (citrate) does improve cancer: A case of a patient suffering from medullary thyroid cancer. Med. Hypotheses 2009, 73, 271. [Google Scholar] [CrossRef] [PubMed]
- Bucay, A.H. Clinical report: A patient with primary peritoneal mesothelioma that has improved after taking citric acid orally. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 241. [Google Scholar] [CrossRef]
- Icard, P.; Fournel, L.; Alifano, M.; Lincet, H. Extracellular Citrate and Cancer Metabolism-Letter. Cancer Res. 2018, 78, 5176. [Google Scholar] [CrossRef] [Green Version]
- Vahidi, R.; Safi, S.; Farsinejad, A.; Panahi, N. Citrate and celecoxib induce apoptosis and decrease necrosis in synergistic manner in canine mammary tumor cells. Cell Mol. Biol. 2015, 61, 22–28. [Google Scholar]
- Cadena, S.M.; Carnieri, E.G.; Echevarria, A.; de Oliveira, M.B. Interference of MI-D, a new mesoionic compound, on artificial and native membranes. Cell Biochem. Funct. 2002, 20, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bigner, D.D.; Velculescu, V.; Parsons, D.W. Mutant metabolic enzymes are at the origin of gliomas. Cancer Res. 2009, 69, 9157–9159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Tesileanu, C.M.S.; Vallentgoed, W.R.; Sanson, M.; Taal, W.; Clement, P.M.; Wick, W.; Brandes, A.A.; Baurain, J.F.; Chinot, O.L.; Wheeler, H.; et al. Non-IDH1-R132H IDH1/2 mutations are associated with increased DNA methylation and improved survival in astrocytomas, compared to IDH1-R132H mutations. Acta Neuropathol. 2021, 141, 945–957. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Rendina, A.R.; Pietrak, B.; Smallwood, A.; Zhao, H.; Qi, H.; Quinn, C.; Adams, N.D.; Concha, N.; Duraiswami, C.; Thrall, S.H.; et al. Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 2013, 52, 4563–4577. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Showalter, M.R.; Hatakeyama, J.; Cajka, T.; VanderVorst, K.; Carraway, K.L.; Fiehn, O. Replication Study: The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Elife 2017, 6, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuyàs, E.; Fernández-Arroyo, S.; Corominas-Faja, B.; Rodríguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; De Llorens, R.; Joven, J.; Menendez, J.A. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Sun, Y.; You, B.; Huang, C.P.; Ye, D.; Chang, C. Androgen receptor reverses the oncometabolite R-2-hydroxyglutarate-induced prostate cancer cell invasion via suppressing the circRNA-51217/miRNA-646/TGFbeta1/p-Smad2/3 signaling. Cancer Lett. 2020, 472, 151–164. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lai, Y.S.; Tsai, H.J.; Kuo, C.C.; Yen, B.L.; Yeh, S.P.; Sun, H.S.; Hung, W.C. The oncometabolite R-2-hydroxyglutarate activates NF-kappaB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci. Rep. 2016, 6, 32428. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.J. (R)-2-hydroxyglutarate or (R)-2HG is an oncometabolite. Bull. Cancer 2013, 100, 655. [Google Scholar] [CrossRef]
- Adam, J.; Yang, M.; Soga, T.; Pollard, P.J. Rare insights into cancer biology. Oncogene 2014, 33, 2547–2556. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Alarcon, T. Metabostemness: A new cancer hallmark. Front. Oncol. 2014, 4, 262. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Semukunzi, H.; Roy, D.; Li, H.; Khan, G.J.; Lyu, X.; Yuan, S.; Lin, S. IDH mutations associated impact on related cancer epidemiology and subsequent effect toward HIF-1alpha. Biomed. Pharmacother. 2017, 89, 805–811. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m (6)A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9. [Google Scholar] [CrossRef]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Cho, H.R.; Yun, T.; Kim, H.; Park, C.K.; Lee, S.H.; Choi, S.H.; Park, S. Metabolomic comparison between cells over-expressing isocitrate dehydrogenase 1 and 2 mutants and the effects of an inhibitor on the metabolism. J. Neurochem. 2015, 132, 183–193. [Google Scholar] [CrossRef]
- Emir, U.E.; Larkin, S.J.; de Pennington, N.; Voets, N.; Plaha, P.; Stacey, R.; Al-Qahtani, K.; Mccullagh, J.; Schofield, C.J.; Clare, S.; et al. Noninvasive Quantification of 2-Hydroxyglutarate in Human Gliomas with IDH1 and IDH2 Mutations. Cancer Res. 2016, 76, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic reprogramming in mutant IDH1 glioma cells. PLoS ONE 2015, 10, e0118781. [Google Scholar] [CrossRef] [Green Version]
- Lenting, K.; Khurshed, M.; Peeters, T.H.; van den Heuvel, C.N.A.M.; van Lith, S.A.M.; de Bitter, T.; Hendriks, W.; Span, P.N.; Molenaar, R.J.; Botman, D.; et al. Isocitrate dehydrogenase 1-mutated human gliomas depend on lactate and glutamate to alleviate metabolic stress. FASEB J. 2019, 33, 557–571. [Google Scholar] [CrossRef]
- Walsby-Tickle, J.; Gannon, J.; Hvinden, I.; Bardella, C.; Abboud, M.I.; Nazeer, A.; Hauton, D.; Pires, E.; Cadoux-Hudson, T.; Schofield, C.J.; et al. Anion-exchange chromatography mass spectrometry provides extensive coverage of primary metabolic pathways revealing altered metabolism in IDH1 mutant cells. Commun. Biol. 2020, 3, 247. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Ohka, F.; Ito, M.; Ranjit, M.; Senga, T.; Motomura, A.; Motomura, K.; Saito, K.; Kato, K.; Kato, Y.; Wakabayashi, T.; et al. Quantitative metabolome analysis profiles activation of glutaminolysis in glioma with IDH1 mutation. Tumour. Biol. 2014, 35, 5911–5920. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.C.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2019, 18, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Hujber, Z.; Horváth, G.; Petővári, G.; Krencz, I.; Dankó, T.; Mészáros, K.; Rajnai, H.; Szoboszlai, N.; Leenders, W.P.J.; Jeney, A.; et al. GABA, glutamine, glutamate oxidation and succinic semialdehyde dehydrogenase expression in human gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 271. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Wu, N.; Li, R.; Zhang, H.; Zhao, Y.; Nie, Y.; Wu, J. IDH2, a novel target of OGT, facilitates glucose uptake and cellular bioenergy production via NF-kappaB signaling to promote colorectal cancer progression. Cell Oncol. 2022, 46, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J.F. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165–49177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [Green Version]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Vatrinet, R.; Leone, G.; De Luise, M.; Girolimetti, G.; Vidone, M.; Gasparre, G.; Porcelli, A.M. The alpha-ketoglutarate dehydrogenase complex in cancer metabolic plasticity. Cancer Metab. 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Dekker, L.J.M.; Verheul, C.; Wensveen, N.; Leenders, W.; Lamfers, M.L.M.; Leenstra, S.; Luider, T.M. Effects of the IDH1 R132H Mutation on the Energy Metabolism: A Comparison between Tissue and Corresponding Primary Glioma Cell Cultures. ACS Omega 2022, 7, 3568–3578. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Cai, L.; Radoul, M.; Chaumeil, M.M.; Blough, M.; Luchman, H.A.; Weiss, S.; Cairncross, J.G.; et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015, 75, 2999–3009. [Google Scholar] [CrossRef] [Green Version]
- Garrett, M.; Sperry, J.; Braas, D.; Yan, W.; Le, T.M.; Mottahedeh, J.; Ludwig, K.; Eskin, A.; Qin, Y.; Levy, R.; et al. Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab. 2018, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, L.J.M.; Wu, S.; Jurriëns, C.; Mustafa, D.A.N.; Grevers, F.; Burgers, P.C.; Sillevis Smitt, P.A.E.; Kros, J.M.; Luider, T.M. Metabolic changes related to the IDH1 mutation in gliomas preserve TCA-cycle activity: An investigation at the protein level. FASEB J. 2020, 34, 3646–3657. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Garcia, J.L.; Cai, L.M.; Chaumeil, M.M.; Eriksson, P.; Robinson, A.E.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Glioma cells with the IDH1 mutation modulate metabolic fractional flux through pyruvate carboxylase. PLoS ONE 2014, 9, e108289. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Chen, Z.; Huang, C.; Shen, J.; Zeng, W.; Feng, S.; Liu, Y.; Li, S.; Chen, M. Development of a novel glycolysis-related genes signature for isocitrate dehydrogenase 1-associated glioblastoma multiforme. Front. Immunol. 2022, 13, 950917. [Google Scholar] [CrossRef]
- van Noorden, C.J.F.; Hira, V.V.V.; van Dijck, A.J.; Novak, M.; Breznik, B.; Molenaar, R.J. Energy Metabolism in IDH1 Wild-Type and IDH1-Mutated Glioblastoma Stem Cells: A Novel Target for Therapy? Cells 2021, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Hohmann, T.; Güttler, A.; Petrenko, M.; Ostheimer, C.; Hohmann, U.; Bache, M.; Dehghani, F.; Vordermark, D. Radiosensitization and a Less Aggressive Phenotype of Human Malignant Glioma Cells Expressing Isocitrate Dehydrogenase 1 (IDH1) Mutant Protein: Dissecting the Mechanisms. Cancers 2019, 11, 889. [Google Scholar] [CrossRef] [Green Version]
- Masson, N.; Ratcliffe, P.J. Hypoxia signaling pathways in cancer metabolism: The importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Bravo-San Pedro, J.M.; Pietrocola, F.; Izzo, V.; Maiuri, M.C.; Kroemer, G.; Galluzzi, L. Assessment of Glycolytic Flux and Mitochondrial Respiration in the Course of Autophagic Responses. Methods Enzymol. 2017, 588, 155–170. [Google Scholar] [PubMed]
- Sica, V.; Bravo-San Pedro, J.M.; Izzo, V.; Pol, J.; Pierredon, S.; Enot, D.; Durand, S.; Bossut, N.; Chery, A.; Souquere, S.; et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition plus Dimethyl alpha-Ketoglutarate. Cell Rep. 2019, 27, 820–834.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, V.; Bravo-San, P.J.; Kroemer, G. A strategy for poisoning cancer cell metabolism: Inhibition of oxidative phosphorylation coupled to anaplerotic saturation. Int. Rev. Cell Mol. Biol. 2019, 347, 27–37. [Google Scholar]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349.e9. [Google Scholar] [CrossRef]
- Sperry, J.; Condro, M.C.; Guo, L.; Braas, D.; Vanderveer-Harris, N.; Kim, K.K.O.; Pope, W.B.; Divakaruni, A.S.; Lai, A.; Christofk, H.; et al. Glioblastoma Utilizes Fatty Acids and Ketone Bodies for Growth Allowing Progression during Ketogenic Diet Therapy. iScience 2020, 23, 101453. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Cuperlovic-Culf, M.; Ferguson, D.; Culf, A.; Morin, P., Jr.; Touaibia, M. 1H NMR metabolomics analysis of glioblastoma subtypes: Correlation between metabolomics and gene expression characteristics. J. Biol. Chem. 2012, 287, 20164–20175. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, H.; Lee, J.H.; Park, J.W. Oxalomalate suppresses metastatic melanoma through IDH-targeted stress response to ROS. Free Radic. Res. 2019, 53, 418–429. [Google Scholar] [CrossRef]
- Erol, A. Type 2 diabetes and cancer as redox diseases? Lancet 2014, 384, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Erol, A. Systemic DNA damage response and metabolic syndrome as a premalignant state. Curr. Mol. Med. 2010, 10, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Ye, S.; Feng, H.; Zeng, C.; Dong, X.; Zeng, X.; Zeng, L.; Lin, X.; Liu, Q.; Yao, J. Silybin suppresses ovarian cancer cell proliferation by inhibiting isocitrate dehydrogenase 1 activity. Cancer Sci. 2022, 113, 3032–3043. [Google Scholar] [CrossRef] [PubMed]
- Kappler, M.; Pabst, U.; Weinholdt, C.; Taubert, H.; Rot, S.; Kaune, T.; Kotrba, J.; Porsch, M.; Güttler, A.; Bache, M.; et al. Causes and Consequences of a Glutamine Induced Normoxic HIF1 Activity for the Tumor Metabolism. Int. J. Mol. Sci. 2019, 20, 4742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Burr, S.P.; Costa, A.S.; Grice, G.L.; Timms, R.T.; Lobb, I.T.; Freisinger, P.; Dodd, R.B.; Dougan, G.; Lehner, P.J.; Frezza, C.; et al. Mitochondrial Protein Lipoylation and the 2-Oxoglutarate Dehydrogenase Complex Controls HIF1alpha Stability in Aerobic Conditions. Cell Metab. 2016, 24, 740–752. [Google Scholar] [CrossRef] [Green Version]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Hansen, G.E.; Gibson, G.E. The alpha-Ketoglutarate Dehydrogenase Complex as a Hub of Plasticity in Neurodegeneration and Regeneration. Int. J. Mol. Sci. 2022, 23, 12403. [Google Scholar] [CrossRef]
- You, Q.; Wang, J.; Yu, Y.; Li, F.; Meng, L.; Chen, M.; Yang, Q.; Xu, Z.; Sun, J.; Zhuo, W.; et al. The histone deacetylase SIRT6 promotes glycolysis through the HIF-1alpha/HK2 signaling axis and induces erlotinib resistance in non-small cell lung cancer. Apoptosis 2022, 27, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhao, Y.; Pan, L.C.; Wang, J.; Shi, X.J.; Du, G.S.; He, Q. Sirolimus increases the anti-cancer effect of Huai Er by regulating hypoxia inducible factor-1alpha-mediated glycolysis in hepatocellular carcinoma. World J. Gastroenterol. 2022, 28, 4600–4619. [Google Scholar] [CrossRef] [PubMed]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.; et al. HIF-1alpha-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [Green Version]
- Williams, N.C.; Ryan, D.G.; Costa, A.S.H.; Mills, E.L.; Jedrychowski, M.P.; Cloonan, S.M.; Frezza, C.; O’Neill, L.A. Signaling metabolite L-2-hydroxyglutarate activates the transcription factor HIF-1alpha in lipopolysaccharide-activated macrophages. J. Biol. Chem. 2022, 298, 101501. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durán, R.V.; MacKenzie, E.D.; Boulahbel, H.; Frezza, C.; Heiserich, L.; Tardito, S.; Bussolati, O.; Rocha, S.; Hall, M.N.; Gottlieb, E. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 2013, 32, 4549–4556. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef]
- Yin, K.; Lee, J.; Liu, Z.; Kim, H.; Martin, D.R.; Wu, D.; Liu, M.; Xue, X. Mitophagy protein PINK1 suppresses colon tumor growth by metabolic reprogramming via p53 activation and reducing acetyl-CoA production. Cell Death Differ. 2021, 28, 2421–2435. [Google Scholar] [CrossRef]
- Lue, H.W.; Podolak, J.; Kolahi, K.; Cheng, L.; Rao, S.; Garg, D.; Xue, C.H.; Rantala, J.K.; Tyner, J.W.; Thornburg, K.L.; et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev. 2017, 31, 2067–2084. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, A.R.; Konig, H.; Johnson, D.E.; Tang, D.; Amaravadi, R.K.; Boyiadzis, M.; Lotze, M.T. You eat what you are: Autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015, 29, 517–525. [Google Scholar] [CrossRef]
- Liu, L.; Li, L.; Li, M.; Luo, Z. Autophagy-Dependent Ferroptosis as a Therapeutic Target in Cancer. ChemMedChem 2021, 16, 942–2950. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Amaravadi, R.K. Emerging strategies to effectively target autophagy in cancer. Oncogene 2016, 35, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, G.; Zhou, Y.; Chen, Y.; Ouyang, L.; Liu, B. Mechanisms of autophagy and relevant small-molecule compounds for targeted cancer therapy. Cell Mol. Life Sci. 2018, 75, 1803–1826. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, L.; Fan, Z.; Xue, W.; Shen, Z.; Yuan, Y.; Sun, X.; Wang, D.; Lian, J.; Wang, L.; et al. Hypoxia-induced GBE1 expression promotes tumor progression through metabolic reprogramming in lung adenocarcinoma. Signal Transduct. Target. Ther. 2020, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Huangyang, P.; Li, F.; Lee, P.; Nissim, I.; Weljie, A.M.; Mancuso, A.; Li, B.; Keith, B.; Yoon, S.S.; Simon, M.C. Fructose-1,6-Bisphosphatase 2 Inhibits Sarcoma Progression by Restraining Mitochondrial Biogenesis. Cell Metab. 2020, 31, 174–188.e7. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4782. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.H.; Jiang, Z.H.; Yang, H.M.; Zhang, Y.; Xu, L.H. Hypoxia-induced FOXO4/LDHA axis modulates gastric cancer cell glycolysis and progression. Clin. Transl. Med. 2021, 11, e279. [Google Scholar] [CrossRef]
- Li, Y.; Lin, S.; Li, L.; Tang, Z.; Hu, Y.; Ban, X.; Zeng, T.; Zhou, Y.; Zhu, Y.; Gao, S.; et al. PDSS2 Deficiency Induces Hepatocarcinogenesis by Decreasing Mitochondrial Respiration and Reprogramming Glucose Metabolism. Cancer Res. 2018, 78, 4471–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network, Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef] [PubMed] [Green Version]
- Li, C.; Tian, C.; Liu, Y.; Liang, J.; Zeng, Y.; Yang, Q.; Liu, Y.; Wu, D.; Wu, J.; Wang, J.; et al. Comprehensive Profiling Reveals Distinct Microenvironment and Metabolism Characterization of Lung Adenocarcinoma. Front. Genet. 2021, 12, 619821. [Google Scholar] [CrossRef]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect Med. 2016, 6, a026146. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Shen, L.; Sun, X.; Fu, Z.; Yang, G.; Li, J.; Yao, L. The fundamental role of the p53 pathway in tumor metabolism and its implication in tumor therapy. Clin. Cancer Res. 2012, 18, 1561–1567. [Google Scholar] [CrossRef] [Green Version]
- García-Cao, I.; García-Cao, M.; Martín-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef] [Green Version]
- Matheu, A.; Maraver, A.; Klatt, P.; Flores, I.; Garcia-Cao, I.; Borras, C.; Flores, J.M.; Viña, J.; Blasco, M.A.; Serrano, M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007, 448, 375–379. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Moreno, M.; Moreno-Cugnon, L.; Matheu, A. Increased Arf/p53 activity in stem cells, aging and cancer. Aging Cell 2017, 16, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Mendrysa, S.M.; O’Leary, K.A.; McElwee, M.K.; Michalowski, J.; Eisenman, R.N.; Powell, D.A.; Perry, M.E. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006, 20, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. Shaping genetic alterations in human cancer: The p53 mutation paradigm. Cancer Cell 2007, 12, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e8. [Google Scholar] [CrossRef] [Green Version]
- Goronzy, J.J.; Fang, F.; Cavanagh, M.M.; Qi, Q.; Weyand, C.M. Naive T cell maintenance and function in human aging. J. Immunol. 2015, 194, 4073–4080. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, A.; Fang, V.; Chen, C.; Serasinghe, M.; Verma, A.; Muller, J.; Chaluvadi, V.S.; Dustin, M.L.; Hla, T.; Elemento, O.; et al. Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature 2017, 546, 158–161. [Google Scholar] [CrossRef] [Green Version]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, S.; Binh Giang, T.; Kan, A.; Marchingo, J.M.; Lye, B.K.; Corcoran, L.M.; Hodgkin, P.D. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat. Immunol. 2017, 18, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasenko, T.N.; Pacheco, S.E.; Koenig, M.K.; Gomez-Rodriguez, J.; Kapnick, S.M.; Diaz, F.; Zerfas, P.M.; Barca, E.; Sudderth, J.; DeBerardinis, R.J.; et al. Cytochrome c Oxidase Activity Is a Metabolic Checkpoint that Regulates Cell Fate Decisions During T Cell Activation and Differentiation. Cell Metab. 2017, 25, 1254–1268.e7. [Google Scholar] [CrossRef] [Green Version]
- Geltink, R.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Qu, G.; Yu, X.; Jiang, H.; Teng, X.L.; Ding, L.; Hu, Q.; Guo, X.; Zhou, Y.; Wang, F.; et al. Acylglycerol Kinase Maintains Metabolic State and Immune Responses of CD8(+) T Cells. Cell Metab. 2019, 30, 290–302.e5. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 2018, 23, 255–269.e4. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, L.; Chen, X.; Shen, Y.; Lian, G.; Shah, N.; Davidoff, A.M.; Yang, J.; Wang, R. MYCN drives glutaminolysis in neuroblastoma and confers sensitivity to an ROS augmenting agent. Cell Death Dis. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Somarribas, P.L.; Vardhana, S.A. Metabolic regulation of the cancer-immunity cycle. Trends Immunol. 2021, 42, 975–993. [Google Scholar] [CrossRef]
- Tigano, M.; Vargas, D.C.; Tremblay-Belzile, S.; Fu, Y.; Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 2021, 591, 477–481. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.e15. [Google Scholar] [CrossRef]
- Caronni, N.; Simoncello, F.; Stafetta, F.; Guarnaccia, C.; Ruiz-Moreno, J.S.; Opitz, B.; Galli, T.; Proux-Gillardeaux, V.; Benvenuti, F. Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer. Cancer Res. 2018, 78, 1685–1699. [Google Scholar] [CrossRef] [Green Version]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic cell reprogramming by endogenously produced lactic acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Mendler, A.N.; Hu, B.; Prinz, P.U.; Kreutz, M.; Gottfried, E.; Noessner, E. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int. J. Cancer 2012, 131, 633–640. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Williams, M.A.; Bevan, M.J. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007, 25, 171–192. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int. J. Cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Kareva, I.; Hahnfeldt, P. The emerging “hallmarks” of metabolic reprogramming and immune evasion: Distinct or linked? Cancer Res. 2013, 73, 2737–2742. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; DeNardo, D.G.; Affara, N.I.; Coussens, L.M. Lymphocytes in cancer development: Polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 2010, 21, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Mednis, A.; Golde, D.W.; David, J.R.; Remold, H.R. Immune (gamma) interferon produced by a human T-lymphoblast cell line. Nature 1981, 292, 842–844. [Google Scholar]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e7. [Google Scholar] [CrossRef] [Green Version]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef]
- Isaacson, B.; Mandelboim, O. Sweet Killers: NK Cells Need Glycolysis to Kill Tumors. Cell Metab. 2018, 28, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255.e5. [Google Scholar] [CrossRef] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jena, B.C.; Das, C.K.; Banerjee, I.; Bharadwaj, D.; Majumder, R.; Das, S.; Biswas, A.; Kundu, M.; Roy, P.K.; Kundu, C.N.; et al. TGF-β1 induced autophagy in cancer associated fibroblasts during hypoxia contributes EMT and glycolysis via MCT4 upregulation. Exp. Cell Res. 2022, 417, 113195. [Google Scholar] [CrossRef]
- Wagner, E.F. Cancer: Fibroblasts for all seasons. Nature 2016, 530, 42–43. [Google Scholar] [CrossRef] [Green Version]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKbeta acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef] [Green Version]
- Utley, A.; Chavel, C.; Lightman, S.; Holling, G.A.; Cooper, J.; Peng, P.; Liu, W.; Barwick, B.G.; Gavile, C.M.; Maguire, O.; et al. CD28 Regulates Metabolic Fitness for Long-Lived Plasma Cell Survival. Cell Rep. 2020, 31, 107815. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Zhou, Y.; Skaro, M.F.; Wu, Y.; Qu, Z.; Mao, F.; Zhao, S.; Xu, Y. Metabolic Reprogramming in Cancer Is Induced to Increase Proton Production. Cancer Res. 2020, 80, 1143–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantelmo, A.R.; Pircher, A.; Kalucka, J.; Carmeliet, P. Vessel pruning or healing: Endothelial metabolism as a novel target? Expert Opin. Ther. Targets 2017, 21, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The history of the angiogenic switch concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Chappell, J.C.; Wiley, D.M.; Bautch, V.L. Regulation of blood vessel sprouting. Semin. Cell Dev. Biol. 2011, 22, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Huang, Y.; Zhu, L.; Yang, K.; Liang, K.; Tan, J.; Yu, B. SIRT6 promotes angiogenesis and hemorrhage of carotid plaque via regulating HIF-1alpha and reactive oxygen species. Cell Death Dis. 2021, 12, 77. [Google Scholar] [CrossRef]
- Huang, Y.H.; Kuo, C.H.; Peng, I.C.; Chang, Y.S.; Tseng, S.H.; Conway, E.M.; Wu, H.L. Recombinant thrombomodulin domain 1 rescues pathological angiogenesis by inhibition of HIF-1alpha-VEGF pathway. Cell Mol. Life Sci. 2021, 78, 7681–7692. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Phng, L.K.; Gerhardt, G. Angiogenesis: A team effort coordinated by notch. Dev. Cell 2009, 16, 196–208. [Google Scholar] [CrossRef]
- Melincovici, C.S.; Boşca, A.B.; Şuşman, S.; Mărginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF)–Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Zhou, H.; Binmadi, N.O.; Yang, Y.H.; Proia, P.; Basile, J.R. Retraction Note to: Semaphorin 4D cooperates with VEGF to promote angiogenesis and tumor progression. Angiogenesis 2020, 23, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–12234. [Google Scholar] [CrossRef] [PubMed]
- Cruys, B.; Wong, B.W.; Kuchnio, A.; Verdegem, D.; Cantelmo, A.R.; Conradi, L.C.; Vandekeere, S.; Bouché, A.; Cornelissen, I.; Vinckier, S.; et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat. Commun. 2016, 7, 2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Mu, P. Targeting Breast Cancer Metastasis. Breast Cancer 2015, 9 (Suppl. S1), 23–34. [Google Scholar] [CrossRef] [Green Version]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, D.K.; Bhattacharya, A.; Mirzaei, R.; Rawji, K.S.; Ahn, Y.; Rho, J.M.; Yong, V.W. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J. Clin. Investig. 2019, 129, 3277–3292. [Google Scholar] [CrossRef] [Green Version]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 2018, 48, 831–832. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Azambuja, A.P.; Simoes-Costa, M. Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 2020, 53, 199–211.e6. [Google Scholar] [CrossRef]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.F.; Chen, S.; Tseng, L.M.; Lee, H.C. Role of the mitochondrial stress response in human cancer progression. Exp. Biol. Med. 2020, 245, 861–878. [Google Scholar] [CrossRef]
- Meng, F.; Wu, L.; Dong, L.; Mitchell, A.V.; James Block, C.; Liu, J.; Zhang, H.; Lu, Q.; Song, W.M.; Zhang, B.; et al. EGFL9 promotes breast cancer metastasis by inducing cMET activation and metabolic reprogramming. Nat. Commun. 2019, 10, 5033. [Google Scholar] [CrossRef] [Green Version]
- Payen, V.L.; Porporato, P.E.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol. Life Sci. 2016, 73, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Le Grand, M.; Mukha, A.; Püschel, J.; Valli, E.; Kamili, A.; Vittorio, O.; Dubrovska, A.; Kavallaris, M. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics 2020, 10, 6411–6429. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Cao, Y.; Meng, G.; Qian, L.; Xu, T.; Yan, C.; Luo, O.; Wang, S.; Wei, J.; Ding, Y.; et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine 2019, 39, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawan, M.A.; Kyrou, I.; Ramanjaneya, M.; Williams, K.; Jeyaneethi, J.; Randeva, H.S.; Karteris, E. Involvement of the glutamine RFamide peptide and its cognate receptor GPR103 in prostate cancer. Oncol. Rep. 2019, 41, 1140–1150. [Google Scholar]
- Mestre-Farrera, A.; Bruch-Oms, M.; Peña, R.; Rodríguez-Morató, J.; Alba-Castellón, L.; Comerma, L.; Quintela-Fandino, M.; Duñach, M.; Baulida, J.; Pozo, Ó.J.; et al. Glutamine-Directed Migration of Cancer-Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res. 2021, 81, 438–451. [Google Scholar] [CrossRef]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Glucose starvation induces LKB1-AMPK-mediated MMP-9 expression in cancer cells. Sci. Rep. 2018, 8, 10122. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Jeong, E.K.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget 2016, 7, 7925–7939. [Google Scholar] [CrossRef] [Green Version]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wei, L.; Lin, G.; Tong, Y.; Zhang, J.; Jiang, X.; He, Q.; Lu, X.; Zhu, D.D.; Chen, Y.Q. Metabolic Alterations Induced by Kudingcha Lead to Cancer Cell Apoptosis and Metastasis Inhibition. Nutr. Cancer 2020, 72, 696–707. [Google Scholar] [CrossRef]
- Liu, H.; Takagaki, Y.; Kumagai, A.; Kanasaki, K.; Koya, D. The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1alpha accumulation. J. Diabetes Investig. 2021, 12, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Draoui, N.; de Zeeuw, P.; Carmeliet, P. Angiogenesis revisited from a metabolic perspective: Role and therapeutic implications of endothelial cell metabolism. Open Biol. 2017, 7, 170219. [Google Scholar] [CrossRef] [Green Version]







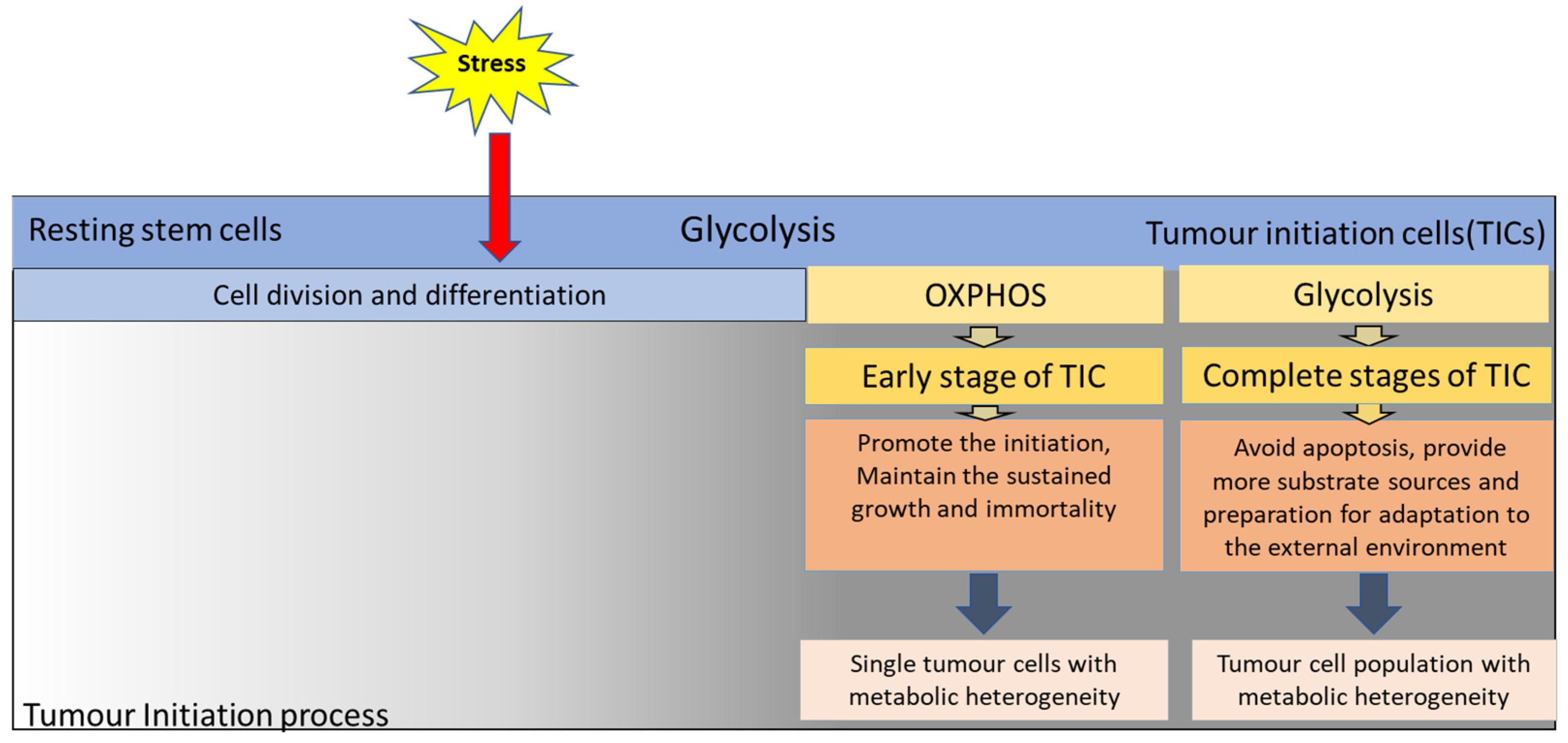{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Medication Conditions | Mechanism |
|---|---|---|
| Cisplatin | HK2 shRNA | Deletion or decrease of HK-2 could produce more ROS, pro-apoptotic molecules, and mitochondrial dysfunction, reduce the drug resistance of tumour cells [154,155,156]. |
| Metformin + sorafenib | HK2 deletion | |
| Clotrimazole | VDAC disruption | The use of PFK inhibitor clotrimazole can reduce glycolysis under the condition of VDAC disruption, prevent the metabolic rearrangement process, and inhibit the growth and invasion of tumour cells [157]. |
| HrDNase1 | DNASE1L3 low expression | HrDNase1 could increase the level of DNASE1L3, then activate the P53-apoptosis pathway, thus inhibiting tumour growth, invasion, and migration [169]. |
| Cetuximab | TRAP1 inhibition | The inhibition of TRAP1 could reduce its binding with PFK1, then trigger the degradation of PFK1, enhancing the sensitivity of CRCs to cetuximab [163]. |
| DASA + metformin | non-sugar environment | DASA inhibits the invasion of tumour cells caused by PKM2 nuclear localisation, and metformin inhibits COXI activity of tumour cells, reduces the growth and promotion of tumour cells [187]. |
| MLN4924 + shikonin + metformin | Tumour cells with high levels of both OXPHOS and glycolysis | MLN4924 inhibits TCA level, shikonin inhibits PKM2 mediated glycolysis, and metformin was used for drug intervention [175,189]. |
| MI-D | DMEM GAL medium | Citric acid could decrease the level of anti-apoptotic molecule expression and increase the level of apoptosis, initiating and executing molecule expression. Then, tumour cells become more sensitive to anti-tumour drugs [210,212,218]. |
| Citric acid + 3-bromo-pyruvate, cisplatin, or celecoxib | — | |
| Doxorubicin + Sorafenib | Insensitive to drugs for mutant IDH1 | The risk scores of glycolysis-related genes were correlated with those two drugs and had better sensitivity [268]. |
| Normoxia + radiotherapy and chemotherapy | tumour cells have the potential of OXPHOS and glycolysis | Remove the cells from the hypoxic area, and then use the corresponding radiotherapy and chemotherapy methods, which can produce more ROS [274,279]. |
| BAY 87-2243(B87) + DMKG | B87 alone is not very effective in inducing tumour cell death | DMKG can produce a strong tumour-killing effect together with B87 [274]. |
| JHU-083 | — | Inhibition of α-KG [276]. |
| Metabolic Type | Factors that Affecting Metabolic Plasticity | Tumour Cell Types | Significance |
|---|---|---|---|
| From Glycolysis to OXPHOS | Resting stem cells are more susceptible to external stimuli during division and differentiation, resulting in increased gene instability, tumorigenesis initiation, and the appearance of tumour-initiating cells (TICs) | The early stage of TIC | For the immortalization and further transformation of TIC by promoting aspartate synthesis and subsequent nucleotide metabolism and other biochemical reactions [83,84,85,86]. |
| Be inclined to Glycolysis | The complete stage of TIC | Avoid apoptosis, and provide more substrate sources and preparation for adaption to the external environment [58,59]. | |
| OXPHOS | With sufficient oxygen | Glioma stem cells (CSCs) | To adapt to different microenvironments and meet their vigorous growth and proliferation [117,118,119,120]. |
| Be inclined to Glycolysis | In the hypoxic regions | ||
| Be inclined to OXPHOS | HIF-1low/pAMPKhigh | Liver hepatocellular carcinoma (HCC), lung adenocarcinoma (LUAD), breast invasive carcinoma, stomach adenocarcinoma, acute myeloid leukaemia and pancreatic adenocarcinoma (PAC) | Hybrid phenotype contributes to metabolic plasticity, allowing tumour cells to adapt to various microenvironments [136]. |
| Be inclined to Glycolysis | HIF-1high/pAMPKlow | ||
| Hybrid phenotype | HIF-1high/pAMPKhigh | ||
| From Glycolysis to OXPHOS | Deletion of HK-2 | Retinal cells, Gastric Cancer, glioblastoma multiforme | More ROS and apoptosis-promoting molecules produce [142,143,144,145]. |
| From Glycolysis to OXPHOS | Inhibitory of PFK expression | Pancreatic ductal adenocarcinoma (PDAC), non-small cell lung cancer (NSCLC) cells, and colorectal cancer (CRCs) | Provide energy for the vigorous metabolism of tumour cells [157,158,159,162,163]. |
| Be inclined to OXPHOS | Dimer PKM2 | - | Entering the nucleus to participate in the transcription and regulation of many molecules, affecting the process of tumour proliferation, migration, angiogenesis, and death [180,181,182]. |
| Be inclined to Glycolysis | Tetramer PKM2 | Providing energy for tumour growth and proliferation [176,177]. | |
| Be inclined to OXPHOS | Citrate synthase (CS) | - | Increase the burden of mitochondrial, causing apoptosis [208,209,210,211,212,213,214]. |
| Be inclined to OXPHOS | Mutation of Isocitrate Dehydrogenase (IDH) | - | Make the (R) enantiomer of 2-hydroxyglutarate (R)-2HG) generate and release abnormal metabolism of lactate and pyruvic acid [248,249,250,251,252,253,254]. |
| Be inclined to OXPHOS | High expression of FBP | - | Inhibit HIF-1α, interfere with glycolysis, and hinder tumour cell proliferation, invasion, and migration [307]. |
| Be inclined to Glycolysis | FBP2 located in the nucleus with c-Myc | - | limiting the expression of mitochondrial genes and reducing OXPHOS energy supply [308]. |
| Be inclined to Glycolysis | Mutations in succinate dehydrogenase (SDH) and fumarate hydratase (FH) | Hereditary paragangliomas, leiomyomatosis and kidney cancers | Reducing the degradation of HIF and promoting the expression of multiple glycolytic enzyme genes [309,310,311]. |
| From OXPHOS to Glycolysis | inhibition of HSP60 | renal cell carcinoma | Enhance de novo nucleotide synthesis and lipid synthesis [312]. |
| From Glycolysis to OXPHOS | Directly regulating PDSS2 | hepatoma cells | Inhibit foci formation, colony formation in soft agar, and tumour formation in nude mice [313]. |
| OXPHOS | WTP53 | - | Reduce harmful products, such as ROS, and obtain more stable genomic DNA, thus avoiding the initiation of tumorigenesis [318,319,320,321,322,323]. |
| From OXPHOS to Glycolysis | MuTP53 | Promotion of tumour proliferation and growth [319]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, N.; Ye, J.; Hu, Z.; Zhang, J.; Wang, Y. Regulative Roles of Metabolic Plasticity Caused by Mitochondrial Oxidative Phosphorylation and Glycolysis on the Initiation and Progression of Tumorigenesis. Int. J. Mol. Sci. 2023, 24, 7076. https://doi.org/10.3390/ijms24087076
Niu N, Ye J, Hu Z, Zhang J, Wang Y. Regulative Roles of Metabolic Plasticity Caused by Mitochondrial Oxidative Phosphorylation and Glycolysis on the Initiation and Progression of Tumorigenesis. International Journal of Molecular Sciences. 2023; 24(8):7076. https://doi.org/10.3390/ijms24087076
Chicago/Turabian StyleNiu, Nan, Jinfeng Ye, Zhangli Hu, Junbin Zhang, and Yun Wang. 2023. "Regulative Roles of Metabolic Plasticity Caused by Mitochondrial Oxidative Phosphorylation and Glycolysis on the Initiation and Progression of Tumorigenesis" International Journal of Molecular Sciences 24, no. 8: 7076. https://doi.org/10.3390/ijms24087076