
Lipid-Sensing Receptor FFAR4 Modulates Pulmonary Epithelial Homeostasis following Immunogenic Exposures Independently of the FFAR4 Ligand Docosahexaenoic Acid (DHA)

 , and
, and
Abstract
:1. Introduction
2. Results
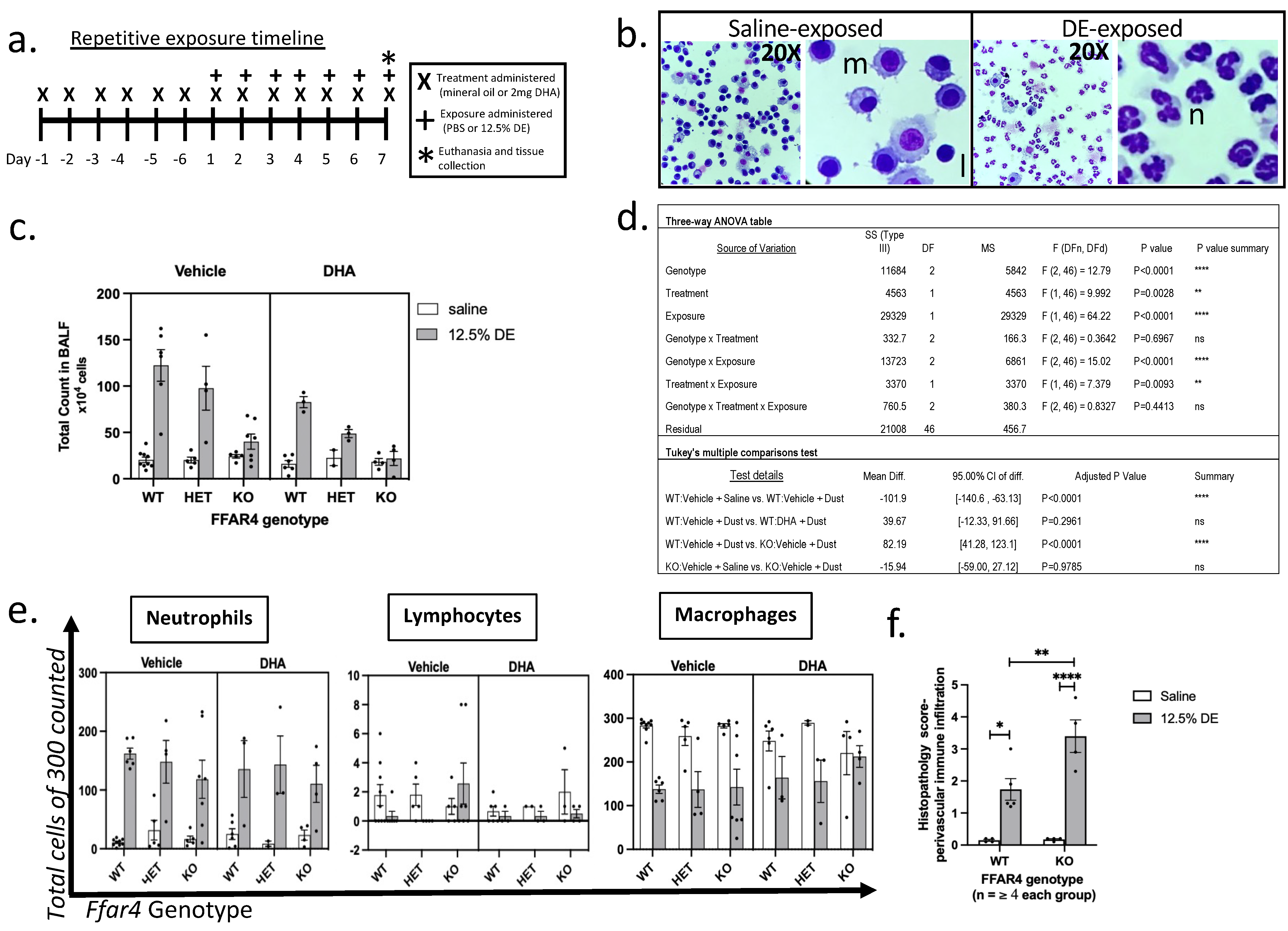
2.1. Ffar4 Deficiency Leads to Dampened Inflammatory Cell Influx to the Airways Independently of DHA
2.2. Dysregulated Epithelial Homeostasis Is Exacerbated by DE Exposure in Ffar4-Null Mice
2.3. Hippo Pathway as a Potential Mechanism of Dysregulated Immune Recruitment and Epithelial Homeostasis
2.4. Genes Related to the Innate Immune Response and Apoptosis Are Downregulated in Ffar4-Deficient Mice
3. Discussion
4. Materials and Methods
4.1. Ffar4 Knockout Mouse Model
4.2. 3R Statement
4.3. Preparation of Hog Barn Dust Extract (DE)
4.4. Dust Exposure Model with Exogenous DHA Administration
4.5. Acute Lung Injury Model
4.6. Parasitic Worm Infection Model
4.7. In Vivo Outcomes
4.8. NanoString nCounter Transcript Analyses
4.9. Histopathological Scoring of FFPE lungs
4.10. Immunofluorescence and Microscopy
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nordgren, T.M.; Friemel, T.D.; Heires, A.J.; Poole, J.A.; Wyatt, T.A.; Romberger, D.J. The omega-3 fatty acid docosahexaenoic acid attenuates organic dust-induced airway inflammation. Nutrients 2014, 6, 5434–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgren, T.M.; Heires, A.J.; Bailey, K.L.; Katafiasz, D.M.; Toews, M.L.; Wichman, C.S.; Romberger, D.J. Docosahexaenoic acid enhances amphiregulin-mediated bronchial epithelial cell repair processes following organic dust exposure. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L421–L431. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Walenta, E. Omega-3 Fatty Acids and FFAR4. Front. Endocrinol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mone, P.; Varzideh, F.; Kansakar, U.; Infante, C.; Lombardi, A.; de Donato, A.; Frullone, S.; Santulli, G. Omega-3 fatty acids coordinate glucose and lipid metabolism in diabetic patients. Lipids Health Dis. 2022, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Skulas-Ray, A.C.; Wilson, P.W.F.; Harris, W.S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G.; et al. Omega-3 Fatty Acids for the Management of Hypertriglyceridemia: A Science Advisory From the American Heart Association. Circulation 2019, 140, e673–e691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elagizi, A.; Lavie, C.J.; O’Keefe, E.; Marshall, K.; O’Keefe, J.H.; Milani, R.V. An Update on Omega-3 Polyunsaturated Fatty Acids and Cardiovascular Health. Nutrients 2021, 13, 204. [Google Scholar] [CrossRef]
- Wood, L.G. Omega-3 polyunsaturated fatty acids and chronic obstructive pulmonary disease. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 128–132. [Google Scholar] [CrossRef]
- Freitas, R.D.S.; Campos, M.M. Understanding the appetite modulation pathways: The role of the FFA1 and FFA4 receptors. Biochem. Pharmacol. 2021, 186, 114503. [Google Scholar] [CrossRef]
- Liao, J.; Xiong, Q.; Yin, Y.; Ling, Z.; Chen, S. The Effects of Fish Oil on Cardiovascular Diseases: Systematical Evaluation and Recent Advance. Front. Cardiovasc. Med. 2021, 8, 802306. [Google Scholar] [CrossRef]
- Dessi, M.; Noce, A.; Bertucci, P.; Manca di Villahermosa, S.; Zenobi, R.; Castagnola, V.; Addessi, E.; Di Daniele, N. Atherosclerosis, dyslipidemia, and inflammation: The significant role of polyunsaturated Fatty acids. ISRN Inflamm. 2013, 2013, 191823. [Google Scholar] [CrossRef] [Green Version]
- Kiepura, A.; Stachyra, K.; Olszanecki, R. Anti-Atherosclerotic Potential of Free Fatty Acid Receptor 4 (FFAR4). Biomedicines 2021, 9, 467. [Google Scholar] [CrossRef] [PubMed]
- So, J.; Wu, D.; Lichtenstein, A.H.; Tai, A.K.; Matthan, N.R.; Maddipati, K.R.; Lamon-Fava, S. EPA and DHA differentially modulate monocyte inflammatory response in subjects with chronic inflammation in part via plasma specialized pro-resolving lipid mediators: A randomized, double-blind, crossover study. Atherosclerosis 2021, 316, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Steffen, B.T.; Podolanczuk, A.J.; Kawut, S.M.; Noth, I.; Raghu, G.; Michos, E.D.; Hoffman, E.A.; Axelsson, G.T.; Gudmundsson, G.; et al. Associations of omega-3 Fatty Acids With Interstitial Lung Disease and Lung Imaging Abnormalities Among Adults. Am. J. Epidemiol. 2021, 190, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chan-Li, Y.; Collins, S.L.; Zhang, Y.; Hallowell, R.W.; Mitzner, W.; Horton, M.R. Pulmonary delivery of docosahexaenoic acid mitigates bleomycin-induced pulmonary fibrosis. BMC Pulm. Med. 2014, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Dyall, S.C.; Balas, L.; Bazan, N.G.; Brenna, J.T.; Chiang, N.; da Costa Souza, F.; Dalli, J.; Durand, T.; Galano, J.M.; Lein, P.J.; et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog. Lipid. Res. 2022, 86, 101165. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Ndreu, R.; Balzan, S. Classes of Lipid Mediators and Their Effects on Vascular Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 1637. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.R.; Edidin, M. Polyunsaturated fatty acids and membrane organization: Elucidating mechanisms to balance immunotherapy and susceptibility to infection. Chem. Phys. Lipids 2008, 153, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, A.; Hasegawa, S.; Kasubuchi, M.; Kimura, I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front. Pharmacol. 2014, 5, 236. [Google Scholar] [CrossRef] [Green Version]
- Milligan, G.; Alvarez-Curto, E.; Hudson, B.D.; Prihandoko, R.; Tobin, A.B. FFA4/GPR120: Pharmacology and Therapeutic Opportunities. Trends Pharmacol. Sci. 2017, 38, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef]
- Lee, K.P.; Park, S.J.; Kang, S.; Koh, J.M.; Sato, K.; Chung, H.Y.; Okajima, F.; Im, D.S. Omega-3 Polyunsaturated fatty acids accelerate airway repair by activating FFA4 in club cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L835–L844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, S.; Hirasawa, A.; Iga, T.; Liu, N.; Itsubo, C.; Sadakane, K.; Hara, T.; Tsujimoto, G. Distribution and regulation of protein expression of the free fatty acid receptor GPR120. Naunyn. Schmiedebergs Arch. Pharmacol. 2009, 379, 427–434. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Occupational Safety and Health. Keeping Farmers Safe. 2020. Available online: https://www.cdc.gov/niosh/newsroom/feature/keepfarmersafe.html (accessed on 19 February 2020).
- May, S.; Romberger, D.J.; Poole, J.A. Respiratory health effects of large animal farming environments. J. Toxicol Environ. Health B Crit. Rev. 2012, 15, 524–541. [Google Scholar] [CrossRef] [Green Version]
- Poole, J.A.; Romberger, D.J. Immunological and inflammatory responses to organic dust in agriculture. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Taskar, V.; Coultas, D. Exposures and idiopathic lung disease. Semin. Respir. Crit. Care Med. 2008, 29, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.M.; Miravitlles, M.; Metzdorf, N.; Celli, B. Impact and prevention of severe exacerbations of COPD: A review of the evidence. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 2891–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrera, M.C.; Labaki, W.W.; Han, M.K. Advances in Chronic Obstructive Pulmonary Disease. Annu. Rev. Med. 2021, 72, 119–134. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Lemoine, C.; Brigham, E.; Woo, H.; Koch, A.; Hanson, C.; Romero, K.; Putcha, N.; McCormack, M.; Hansel, N. Relationship between Omega-3 and Omega-6 Fatty Acid Intake and Chronic Obstructive Pulmonary Disease Morbidity. Ann. Am. Thorac. Soc. 2020, 17, 378–381. [Google Scholar] [CrossRef]
- Bjursell, M.; Xu, X.; Admyre, T.; Bottcher, G.; Lundin, S.; Nilsson, R.; Stone, V.M.; Morgan, N.G.; Lam, Y.Y.; Storlien, L.H.; et al. The beneficial effects of n-3 polyunsaturated fatty acids on diet induced obesity and impaired glucose control do not require Gpr120. PLoS ONE 2014, 9, e114942. [Google Scholar] [CrossRef] [Green Version]
- Paerregaard, S.I.; Agerholm, M.; Serup, A.K.; Ma, T.; Kiens, B.; Madsen, L.; Kristiansen, K.; Jensen, B.A. FFAR4 (GPR120) Signaling Is Not Required for Anti-Inflammatory and Insulin-Sensitizing Effects of Omega-3 Fatty Acids. Mediat. Inflamm. 2016, 2016, 1536047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.N.; Harkema, J.R.; Nelson, A.J.; Dickinson, J.D.; Kalil, J.; Duryee, M.J.; Thiele, G.M.; Kumar, B.; Singh, A.B.; Gaurav, R.; et al. MyD88 regulates a prolonged adaptation response to environmental dust exposure-induced lung disease. Respir. Res. 2020, 21, 97. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Nemecek, M.; Chandra, D.; DeVasure, J.M.; Nelson, A.J.; Romberger, D.J.; Poole, J.A. Organic dust-induced lung injury and repair: Bi-directional regulation by TNFalpha and IL-10. J. Immunotoxicol. 2020, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, T.M.; Bauer, C.D.; Heires, A.J.; Poole, J.A.; Wyatt, T.A.; West, W.W.; Romberger, D.J. Maresin-1 reduces airway inflammation associated with acute and repetitive exposures to organic dust. Transl. Res. 2015, 166, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, M.G.; Herbert, D.R. Immune polarization by hookworms: Taking cues from T helper type 2, type 2 innate lymphoid cells and alternatively activated macrophages. Immunology 2016, 148, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wu, S.; Lu, R.; Zhang, Y.G.; Zheng, Y.; Sun, J. Pulmonary permeability assessed by fluorescent-labeled dextran instilled intranasally into mice with LPS-induced acute lung injury. PLoS ONE 2014, 9, e101925. [Google Scholar] [CrossRef]
- Volckaert, T.; Yuan, T.; Yuan, J.; Boateng, E.; Hopkins, S.; Zhang, J.S.; Thannickal, V.J.; Fassler, R.; De Langhe, S.P. Hippo signaling promotes lung epithelial lineage commitment by curbing Fgf10 and beta-catenin signaling. Development 2019, 146, dev166454. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhou, L.; Ling, L.; Meng, X.; Chu, F.; Zhang, S.; Zhou, F. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front. Immunol. 2020, 11, 323. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Hu, Z.; Qi, H.; Shi, Z.; Chang, Y.; Yao, Q.; Cui, H.; Zheng, L.; Han, Y.; Han, X.; et al. G-protein-coupled receptors mediate omega-3 PUFAs-inhibited colorectal cancer by activating the Hippo pathway. Oncotarget 2016, 7, 58315–58330. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, J.E.; Mori, M.; Szymaniak, A.D.; Varelas, X.; Cardoso, W.V. The hippo pathway effector Yap controls patterning and differentiation of airway epithelial progenitors. Dev. Cell 2014, 30, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Hicks-Berthet, J.; Ning, B.; Federico, A.; Tilston-Lunel, A.; Matschulat, A.; Ai, X.; Lenburg, M.E.; Beane, J.; Monti, S.; Varelas, X. Yap/Taz inhibit goblet cell fate to maintain lung epithelial homeostasis. Cell Rep. 2021, 36, 109347. [Google Scholar] [CrossRef]
- Ulu, A.; Burr, A.; Heires, A.J.; Pavlik, J.; Larsen, T.; Perez, P.A.; Bravo, C.; DiPatrizio, N.V.; Baack, M.; Romberger, D.J.; et al. A high docosahexaenoic acid diet alters lung inflammation and recovery following repetitive exposure to aqueous organic dust extracts. J. Nutr. Biochem. 2021, 97, 108797. [Google Scholar] [CrossRef]
- Larsson, K.A.; Eklund, A.G.; Hansson, L.O.; Isaksson, B.M.; Malmberg, P.O. Swine dust causes intense airways inflammation in healthy subjects. Am. J. Respir. Crit. Care Med. 1994, 150, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Larsson, B.M.; Palmberg, L.; Malmberg, P.O.; Larsson, K. Effect of exposure to swine dust on levels of IL-8 in airway lavage fluid. Thorax 1997, 52, 638–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, E.W.; Palmberg, L. Differential Pattern of Human Blood Neutrophil Activation After Stimulation With Organic Dust in Vitro and in Vivo. J. Occup. Environ. Med. 2007, 49, 131–138. [Google Scholar] [CrossRef] [PubMed]
- McGovern, T.K.; Chen, M.; Allard, B.; Larsson, K.; Martin, J.G.; Adner, M. Neutrophilic oxidative stress mediates organic dust-induced pulmonary inflammation and airway hyperresponsiveness. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L155–L165. [Google Scholar] [CrossRef] [Green Version]
- Von Essen, S.; Romberger, D. The respiratory inflammatory response to the swine confinement building environment: The adaptation to respiratory exposures in the chronically exposed worker. J. Agric. Saf. Health 2003, 9, 185–196. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid mediators in the resolution of inflammation. Cold Spring Harb. Perspect. Biol. 2014, 7, a016311. [Google Scholar] [CrossRef] [Green Version]
- Turk, H.F.; Chapkin, R.S. Membrane lipid raft organization is uniquely modified by n-3 polyunsaturated fatty acids. Prostaglandins Leukot Essent Fat. Acids 2013, 88, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, J.C.; Van Dyken, S.J.; von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Barnes, M.A.; Sureshchandra, S.; Menicucci, A.R.; Patel, J.J.; Messaoudi, I.; Nair, M.G. CX3CR1-Expressing Myeloid Cells Regulate Host-Helminth Interaction and Lung Inflammation. Adv. Biol. 2022, 6, e2101078. [Google Scholar] [CrossRef] [PubMed]
- King, I.L.; Li, Y. Host-Parasite Interactions Promote Disease Tolerance to Intestinal Helminth Infection. Front. Immunol. 2018, 9, 2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbino, F.; Garlatti, V.; Garzarelli, V.; Massimino, L.; Spano, S.; Iadarola, P.; Cagnone, M.; Giera, M.; Heijink, M.; Guglielmetti, S.; et al. GPR120 prevents colorectal adenocarcinoma progression by sustaining the mucosal barrier integrity. Sci. Rep. 2022, 12, 381. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Meier, K.E. Free fatty acid receptor (FFAR) agonists inhibit proliferation of human ovarian cancer cells. Prostaglandins Leukot Essent Fat. Acids 2017, 122, 24–29. [Google Scholar] [CrossRef]
- Kyomoto, Y.; Kanazawa, H.; Tochino, Y.; Watanabe, T.; Asai, K.; Kawaguchi, T. Possible role of airway microvascular permeability on airway obstruction in patients with chronic obstructive pulmonary disease. Respir. Med. 2019, 146, 137–141. [Google Scholar] [CrossRef]
- Carlier, F.M.; de Fays, C.; Pilette, C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front. Physiol. 2021, 12, 691227. [Google Scholar] [CrossRef]
- Sigsgaard, T.; Basinas, I.; Doekes, G.; de Blay, F.; Folletti, I.; Heederik, D.; Lipinska-Ojrzanowska, A.; Nowak, D.; Olivieri, M.; Quirce, S.; et al. Respiratory diseases and allergy in farmers working with livestock: A EAACI position paper. Clin. Transl. Allergy 2020, 10, 29. [Google Scholar] [CrossRef]
- Ooi, D.S.; Tan, C.P.; Tay, M.J.; Ong, S.G.; Tham, E.H.; Siah, K.T.H.; Eriksson, J.G.; Godfrey, K.M.; Shek, L.P.; Loo, E.X. Developmental Origins of Health and Disease: Impact of environmental dust exposure in modulating microbiome and its association with non-communicable diseases. J. Dev. Orig. Health Dis. 2020, 11, 545–556. [Google Scholar] [CrossRef]
- Janbazacyabar, H.; van Bergenhenegouwen, J.; Varasteh, S.; Garssen, J.; Folkerts, G.; Braber, S. Repeated exposure of bronchial epithelial cells to particular matter increases allergen-induced cytokine release and permeability. Cytokine 2022, 154, 155878. [Google Scholar] [CrossRef]
- Shrestha, D.; Massey, N.; Bhat, S.M.; Jelesijevic, T.; Sahin, O.; Zhang, Q.; Bailey, K.L.; Poole, J.A.; Charavaryamath, C. Nrf2 Activation Protects Against Organic Dust and Hydrogen Sulfide Exposure Induced Epithelial Barrier Loss and K. pneumoniae Invasion. Front. Cell Infect. Microbiol. 2022, 12, 848773. [Google Scholar] [CrossRef] [PubMed]
- Huber, D.; Balda, M.S.; Matter, K. Occludin modulates transepithelial migration of neutrophils. J. Biol. Chem. 2000, 275, 5773–5778. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.H.; Cheng, S.L.; Liu, H.T.; Yang, F.Z.; Wang, H.C.; Yu, C.J. Elastase induced lung epithelial cell apoptosis and emphysema through placenta growth factor. Cell Death Dis. 2013, 4, e793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindel, D.; Mensat, P.; Vol, C.; Homayed, Z.; Charrier-Savournin, F.; Trinquet, E.; Baneres, J.L.; Pin, J.P.; Pannequin, J.; Roux, T.; et al. G protein-coupled receptors can control the Hippo/YAP pathway through Gq signaling. FASEB J. 2021, 35, e21668. [Google Scholar] [CrossRef]
- Wang, J.; Hong, Y.; Shao, S.; Zhang, K.; Hong, W. FFAR1-and FFAR4-dependent activation of Hippo pathway mediates DHA-induced apoptosis of androgen-independent prostate cancer cells. Biochem. Biophys. Res. Commun. 2018, 506, 590–596. [Google Scholar] [CrossRef]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.U.; Williams, B.R.; Hannigan, G.E. Transcriptional Activation of Inflammatory Genes: Mechanistic Insight into Selectivity and Diversity. Biomolecules 2015, 5, 3087–3111. [Google Scholar] [CrossRef] [PubMed]
- LeVan, T.D.; Romberger, D.J.; Siahpush, M.; Grimm, B.L.; Ramos, A.K.; Johansson, P.L.; Michaud, T.L.; Heires, A.J.; Wyatt, T.A.; Poole, J.A. Relationship of systemic IL-10 levels with proinflammatory cytokine responsiveness and lung function in agriculture workers. Respir. Res. 2018, 19, 166. [Google Scholar] [CrossRef]
- Muller-Suur, C.; Larsson, P.H.; Larsson, K. T-cell activation by organic dust in vitro. Respir. Med. 2000, 94, 821–827. [Google Scholar] [CrossRef] [Green Version]
- Muller-Suur, C.; Larsson, P.H.; Larsson, K.; Grunewald, J. Lymphocyte activation after exposure to swine dust: A role of humoral mediators and phagocytic cells. Eur. Respir. J. 2002, 19, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Sahlander, K.; Larsson, K.; Palmberg, L. Daily exposure to dust alters innate immunity. PLoS ONE 2012, 7, e31646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Kang, T.B. The Molecular Links between Cell Death and Inflammasome. Cells 2019, 8, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datilo, M.N.; Sant’Ana, M.R.; Formigari, G.P.; Rodrigues, P.B.; de Moura, L.P.; da Silva, A.S.R.; Ropelle, E.R.; Pauli, J.R.; Cintra, D.E. Omega-3 from Flaxseed Oil Protects Obese Mice Against Diabetic Retinopathy Through GPR120 Receptor. Sci. Rep. 2018, 8, 14318. [Google Scholar] [CrossRef] [Green Version]
- Diakogiannaki, E.; Dhayal, S.; Childs, C.E.; Calder, P.C.; Welters, H.J.; Morgan, N.G. Mechanisms involved in the cytotoxic and cytoprotective actions of saturated versus monounsaturated long-chain fatty acids in pancreatic beta-cells. J. Endocrinol. 2007, 194, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Amos, D.; Cook, C.; Santanam, N. Omega 3 rich diet modulates energy metabolism via GPR120-Nrf2 crosstalk in a novel antioxidant mouse model. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 466–488. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, F.; Palmberg, L.; Larsson, K. Exposure to organic dust causes activation of human plasma complement factors C3 and B and the synthesis of factor C3 by lung epithelial cells in vitro. Inflammation 2005, 29, 39–45. [Google Scholar] [CrossRef]
- Pandya, P.H.; Wilkes, D.S. Complement system in lung disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, H.J.; Iversen, M.; Brandslund, I.; Sigsgaard, T.; Omland, O.; Oxvig, C.; Holmskov, U.; Bjermer, L.; Jensenius, J.C.; Dahl, R. Plasma C3d levels of young farmers correlate with respirable dust exposure levels during normal work in swine confinement buildings. Ann. Agric. Environ. Med. 2003, 10, 53–60. [Google Scholar]
- Sin, W.X.; Yeong, J.P.; Lim, T.J.F.; Su, I.H.; Connolly, J.E.; Chin, K.C. IRF-7 Mediates Type I IFN Responses in Endotoxin-Challenged Mice. Front. Immunol. 2020, 11, 640. [Google Scholar] [CrossRef] [Green Version]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem. Cell 2020, 26, 346–358.e4. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Han, J.; Khan, Z.A.; Fang, Q.; Jin, Y.; Chen, X.; Zhang, Y.; Wang, M.; Qian, J.; et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat. Commun. 2020, 11, 2148. [Google Scholar] [CrossRef]
- Poole, J.A.; Wyatt, T.A.; Romberger, D.J.; Staab, E.; Simet, S.; Reynolds, S.J.; Sisson, J.H.; Kielian, T. MyD88 in lung resident cells governs airway inflammatory and pulmonary function responses to organic dust treatment. Respir. Res. 2015, 16, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungurianu, A.; Margina, D.; Gradinaru, D.; Bacanu, C.; Ilie, M.; Tsitsimpikou, C.; Tsarouhas, K.; Spandidos, D.A.; Tsatsakis, A.M. Lipoprotein redox status evaluation as a marker of cardiovascular disease risk in patients with inflammatory disease. Mol. Med. Rep. 2017, 15, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Kotlyarov, S. High-Density Lipoproteins: A Role in Inflammation in COPD. Int. J. Mol. Sci. 2022, 23, 8128. [Google Scholar] [CrossRef]
- Lin, X.; Ma, P.; Yang, C.; Wang, J.; He, K.; Chen, G.; Huang, W.; Fan, J.; Xian, X.; Wang, Y.; et al. Dietary-Induced Elevations of Triglyceride-Rich Lipoproteins Promote Atherosclerosis in the Low-Density Lipoprotein Receptor Knockout Syrian Golden Hamster. Front. Cardiovasc Med. 2021, 8, 738060. [Google Scholar] [CrossRef] [PubMed]
- Adiels, M.; Olofsson, S.O.; Taskinen, M.R.; Boren, J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Ozdener, M.H.; Subramaniam, S.; Sundaresan, S.; Sery, O.; Hashimoto, T.; Asakawa, Y.; Besnard, P.; Abumrad, N.A.; Khan, N.A. CD36- and GPR120-mediated Ca(2)(+) signaling in human taste bud cells mediates differential responses to fatty acids and is altered in obese mice. Gastroenterology 2014, 146, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.A.; Harsch, B.A.; Healy, C.L.; Joshi, S.S.; Huang, S.; Walker, R.E.; Wagner, B.M.; Ernste, K.M.; Huang, W.; Block, R.C.; et al. Free fatty acid receptor 4 responds to endogenous fatty acids to protect the heart from pressure overload. Cardiovasc. Res. 2022, 118, 1061–1073. [Google Scholar] [CrossRef]
- Carullo, G.; Mazzotta, S.; Vega-Holm, M.; Iglesias-Guerra, F.; Vega-Perez, J.M.; Aiello, F.; Brizzi, A. GPR120/FFAR4 Pharmacology: Focus on Agonists in Type 2 Diabetes Mellitus Drug Discovery. J. Med. Chem. 2021, 64, 4312–4332. [Google Scholar] [CrossRef]
- Sorensen, K.V.; Korfitzen, S.S.; Kaspersen, M.H.; Ulven, E.R.; Ekberg, J.H.; Bauer-Brandl, A.; Ulven, T.; Hojlund, K. Acute effects of delayed-release hydrolyzed pine nut oil on glucose tolerance, incretins, ghrelin and appetite in healthy humans. Clin. Nutr. 2021, 40, 2169–2179. [Google Scholar] [CrossRef]
- Romberger, D.J.; Bodlak, V.; Von Essen, S.G.; Mathisen, T.; Wyatt, T.A. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J. Appl. Physiol. 2002, 93, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Romberger, D.J.; Heires, A.J.; Nordgren, T.M.; Poole, J.A.; Toews, M.L.; West, W.W.; Wyatt, T.A. beta2-Adrenergic agonists attenuate organic dust-induced lung inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L101–L110. [Google Scholar] [CrossRef] [Green Version]
- Baggio, C.; Velazquez, J.V.; Fragai, M.; Nordgren, T.M.; Pellecchia, M. Therapeutic Targeting of MMP-12 for the Treatment of Chronic Obstructive Pulmonary Disease. J. Med. Chem. 2020, 63, 12911–12920. [Google Scholar] [CrossRef]
- Hamakawa, H.; Bartolak-Suki, E.; Parameswaran, H.; Majumdar, A.; Lutchen, K.R.; Suki, B. Structure-function relations in an elastase-induced mouse model of emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 45, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batugedara, H.M.; Li, J.; Chen, G.; Lu, D.; Patel, J.J.; Jang, J.C.; Radecki, K.C.; Burr, A.C.; Lo, D.D.; Dillman, A.R.; et al. Hematopoietic cell-derived RELMalpha regulates hookworm immunity through effects on macrophages. J. Leukoc. Biol. 2018, 104, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Velazquez, J.V.; Burr, A.; Sveiven, S.N.; Yang, J.; Bravo, C.; Hammock, B.D.; Nordgren, T.M. Sex-Specific Differences in Resolution of Airway Inflammation in Fat-1 Transgenic Mice Following Repetitive Agricultural Dust Exposure. Front. Pharmacol. 2021, 12, 785193. [Google Scholar] [CrossRef]
- Wu, G.; Haw, R. Functional Interaction Network Construction and Analysis for Disease Discovery. Methods Mol. Biol. 2017, 1558, 235–253. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Dwyer-Nield, L.D.; Kwon, J.B.; Barthel, L.; Janssen, W.J.; Merrick, D.T.; Keith, R.L. Tracheal dysplasia precedes bronchial dysplasia in mouse model of N-nitroso trischloroethylurea induced squamous cell lung cancer. PLoS ONE 2015, 10, e0122823. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Analysis of Unique DEGs from WT DE-Exposed vs. WT Saline Comparison | |
|---|---|
| Reactome Pathway Name | p-Value |
| RUNX1 and FOXP3 control the development of Tregs | 5.13 × 10−5 |
| Chemokine receptors bind chemokines | 6.03 × 10−5 |
| Interleukin-10 signaling | 5.40 × 10−4 |
| Alternative complement activation | 2.24 × 10−3 |
| Activation of C3 and C5 | 2.24 × 10−3 |
| Interleukin-4 and Interleukin-13 signaling | 3.38 × 10−3 |
| Pathway Analysis of Unique DEGs from KO DE-Exposed vs. KO Saline Comparison | |
| Reactome Pathway Name | p-Value |
| CLEC7A/inflammasome pathways | 2.55 × 10−6 |
| Interleukin-10 signaling | 2.74 × 10−5 |
| Interleukin-1 processing | 3.06 × 10−5 |
| Pyroptosis | 4.78 × 10−4 |
| C-type lectin receptors | 1.04 × 10−3 |
| RUNX3 regulates immune response and cell migration | 2.12 × 10−3 |
| Regulated necrosis | 4.76 × 10−3 |
| Dectin-2 family | 7.14 × 10−3 |
| Pathway Analysis of DEGs in DE-Exposed KO vs. WT Mice | |
| Reactome Pathway Name | p-Value |
| Activation of IRF3/IRF7-mediated TBK1/IKK epsilon | 4.66 × 10−4 |
| TRAF6-mediated IRF7 activation of TLR7/8 or 9 signaling | 2.21 × 10−3 |
| TICAM1-dependent activation of IRF3/IRF7 | 4.42 × 10−3 |
| TRAF6-mediated IRF7 activation | 8.81 × 10−3 |
| TRAF3-dependent IRF activation pathway | 8.81 × 10−3 |
| Alternative complement activation | 1.32 × 10−2 |
| Activation of C3 and C5 | 1.32 × 10−2 |
| DEx/H-box helicases activate type I IFN and inflammatory cytokine production | 1.53 × 10−2 |
| MyD88-dependent cascade initiated on endosome | 1.94 × 10−2 |
| Toll Like receptor 7/8 (TLR7/8) cascade | 1.98 × 10−2 |
| TRIF-mediated programmed cell death | 2.19 × 10−2 |
| Toll-like receptor 9 cascade | 2.25 × 10−2 |
| MyD88-independent TLR4 cascade | 2.34 × 10−2 |
| TRIF(TICAM1)-mediated TLR4 signaling | 2.34 × 10−2 |
| TRIF-mediated programmed cell death | 2.41 × 10−2 |
| Toll-like receptor 4 cascade | 3.24 × 10−2 |
| Activation of IRF3/IRF7 mediated by TBK1/IKK epsilon | 3.35 × 10−2 |
| Caspase activation via Death Receptors in the presence of ligand | 3.48 × 10−2 |
| Activation of TAK1 complex upon TLR7/8 or 9 stimulation | 3.82 × 10−2 |
| TRAF6-mediated induction of TAK1 complex within TLR4 complex | 4.00 × 10−2 |
| IRAK deficiency (TLR2/4) | 4.17 × 10−2 |
| IKK complex recruitment mediated by RIP1 | 4.47 × 10−2 |
| Caspase activation via extrinsic apoptotic signaling pathway | 4.69 × 10−2 |
| Heme signaling | 4.91 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sveiven, S.N.; Anesko, K.; Morgan, J.; Nair, M.G.; Nordgren, T.M. Lipid-Sensing Receptor FFAR4 Modulates Pulmonary Epithelial Homeostasis following Immunogenic Exposures Independently of the FFAR4 Ligand Docosahexaenoic Acid (DHA). Int. J. Mol. Sci. 2023, 24, 7072. https://doi.org/10.3390/ijms24087072
Sveiven SN, Anesko K, Morgan J, Nair MG, Nordgren TM. Lipid-Sensing Receptor FFAR4 Modulates Pulmonary Epithelial Homeostasis following Immunogenic Exposures Independently of the FFAR4 Ligand Docosahexaenoic Acid (DHA). International Journal of Molecular Sciences. 2023; 24(8):7072. https://doi.org/10.3390/ijms24087072
Chicago/Turabian StyleSveiven, Stefanie N., Kyle Anesko, Joshua Morgan, Meera G. Nair, and Tara M. Nordgren. 2023. "Lipid-Sensing Receptor FFAR4 Modulates Pulmonary Epithelial Homeostasis following Immunogenic Exposures Independently of the FFAR4 Ligand Docosahexaenoic Acid (DHA)" International Journal of Molecular Sciences 24, no. 8: 7072. https://doi.org/10.3390/ijms24087072