
The Pathogenesis of Diabetes

Abstract
:1. Status and Characteristics of Global Diabetes
2. Diabetes Triggered by Inflammation
3. Autophagy Prevented Diabetes and Its Complications
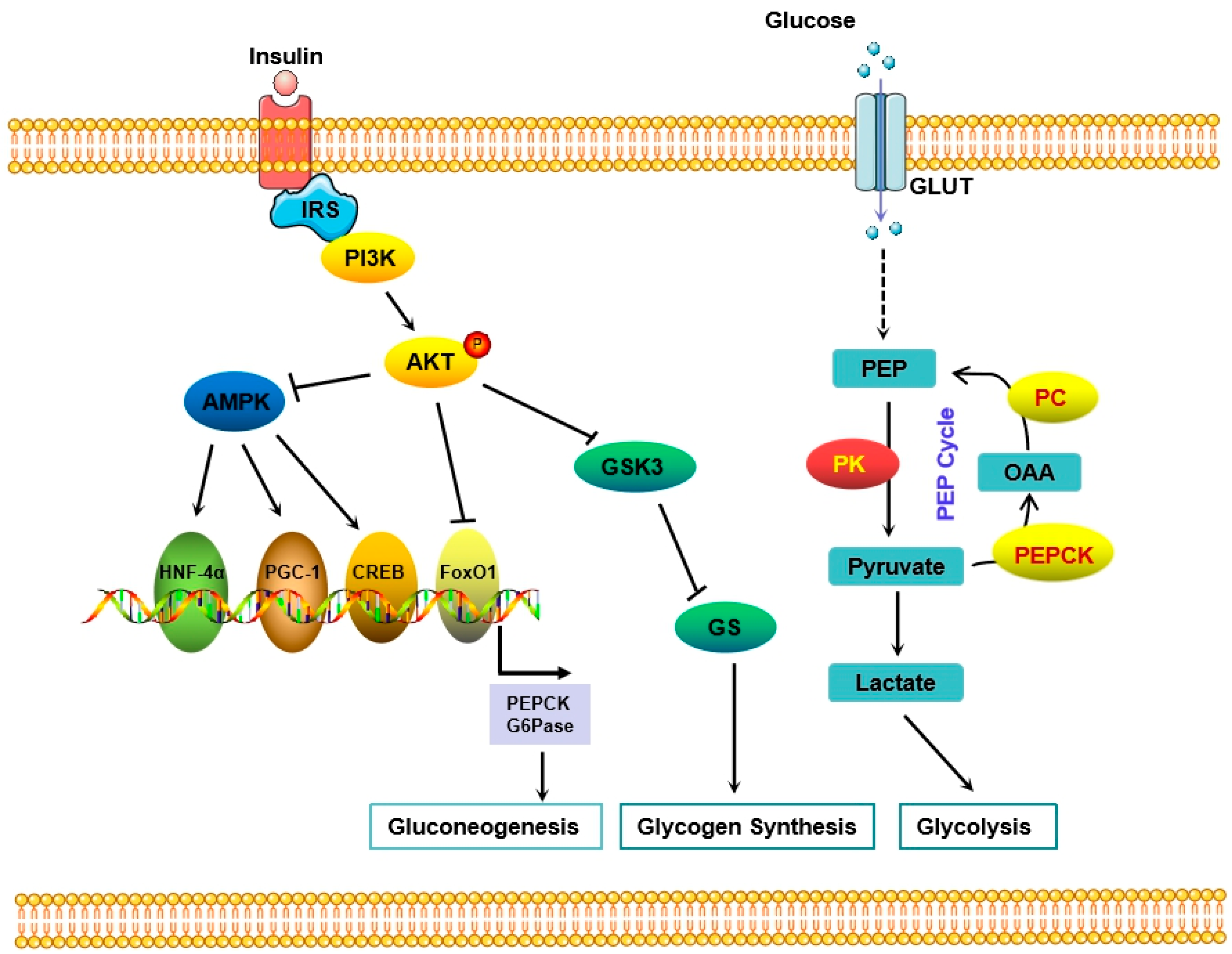
4. Glucose Homeostasis
5. The Role of the Liver in Regulating Glucose Metabolism
5.1. Glucose Transporters
5.2. Glycogen Synthesis
5.3. Gluconeogenesis and Upstream Transcription Factors
6. Insulin-Mediated Signaling Pathway in Diabetes
6.1. The MAPK Pathway Regulates Insulin-Mediated Metabolism
6.2. The PI3K/Akt Pathway Was Involved in Insulin Resistance
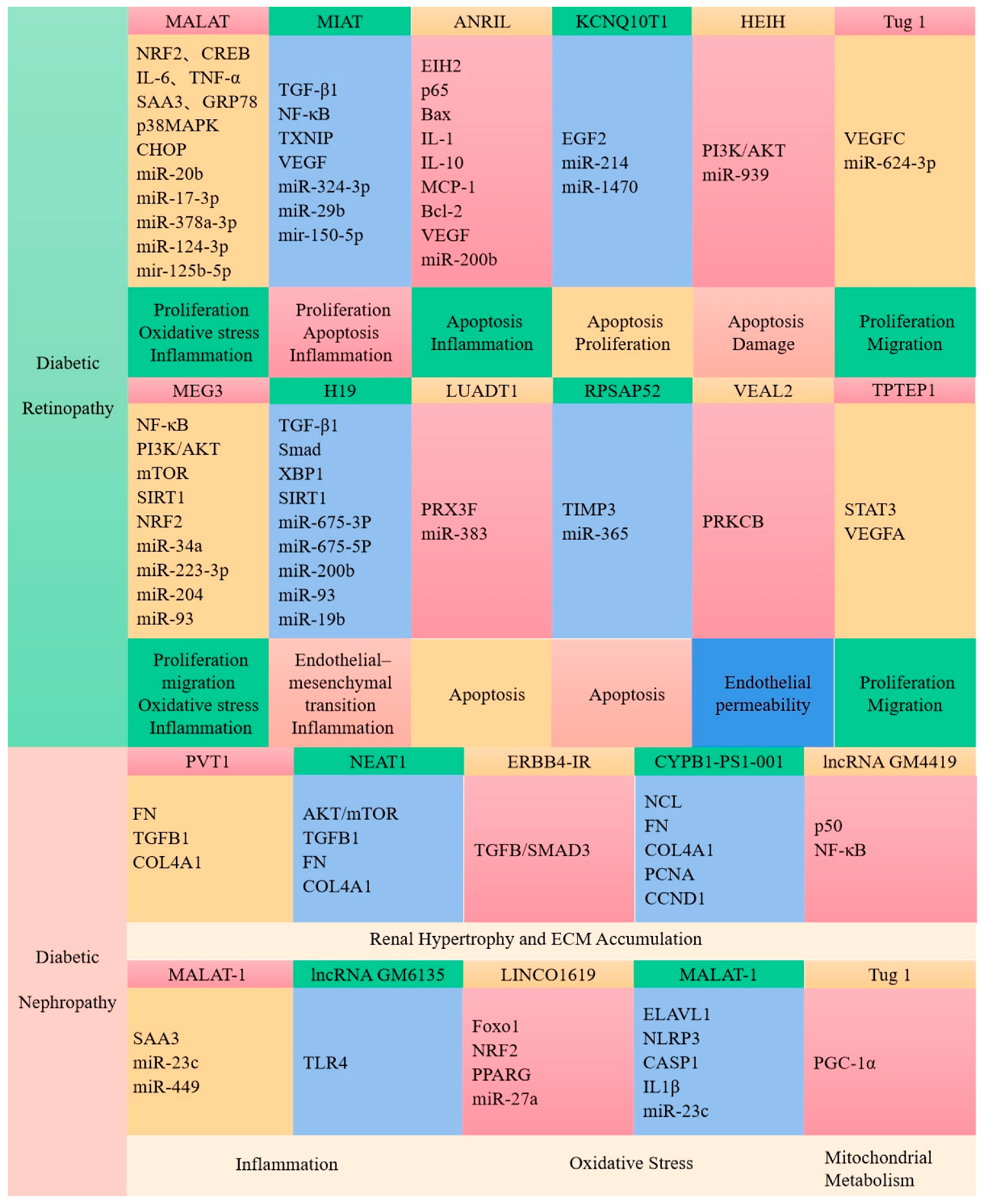
7. Roles of Long Non-Coding RNA in Diabetes and Its Complications
7.1. LncRNAs Regulate Glucose Metabolism in Diabetes
7.2. Roles of LncRNAs in Islet
7.3. LncRNAs Are Associated with Diabetic Complications
8. The Disturbance of Intestinal Flora May Partly Give Rise to Diabetes
9. Sirtuins as a Novel Drug Target for Type 2 Diabetes
10. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. IDF Diabetes Atlas Committee: Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045, Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef]
- Ma, Q.; Li, Y.; Li, P.; Wang, M.; Wang, J.; Tang, Z.; Wang, T.; Luo, L.; Wang, C.; Wang, T.; et al. Research progress in the relationship between type 2 diabetes mellitus and intestinal flora. Biomed. Pharmacother. 2019, 117, 109138. [Google Scholar] [CrossRef]
- Chen, L.; Sun, X.; Xiao, H.; Xu, F.; Yang, Y.; Lin, Z.; Chen, Z.; Quan, S.; Huang, H. PAQR3 regulates phosphorylation of FoxO1 in insulin-resistant HepG2 cells via NF-κB signaling pathway. Exp. Cell. Res. 2019, 381, 301–310. [Google Scholar] [CrossRef]
- Schwartsburd, P. Glucose-lowering Strategies in Diabetes: Pharmacological Development of New Antidiabetic Drugs. Curr. Pharm. Des. 2018, 24, 1007–1011. [Google Scholar] [CrossRef]
- Alsahli, M.; Gerich, J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef]
- Malone, J.I.; Hansen, B.C. Does obesity cause type 2 diabetes mellitus (T2DM)? Or is it the opposite? Pediatr. Diabetes 2019, 20, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, S.W.; Wang, L.J.; Bai, Y.M.; Zeng, X.Y.; Guo, H.B.; Liu, Y.N.; Jiang, Y.Y.; Dong, W.L.; He, G.X.; et al. Burden of diabetes, hyperglycaemia in China from to 2016, Findings from the 1990 to 2016, global burden of disease study. Diabetes Metab. 2019, 45, 286–293. [Google Scholar] [CrossRef]
- Lovic, D.; Piperidou, A.; Zografou, I.; Grassos, H.; Pittaras, A.; Manolis, A. The Growing Epidemic of Diabetes Mellitus. Curr. Vasc. Pharmacol. 2020, 18, 104–109. [Google Scholar] [CrossRef]
- Lekva, T.; Norwitz, E.R.; Aukrust, P.; Ueland, T. Impact of Systemic Inflammation on the Progression of Gestational Diabetes Mellitus. Curr. Diab. Rep. 2016, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; He, Q.; Peng, B.; Chiao, P.J.; Ye, J. Regulation of nuclear translocation of HDAC3 by IkappaBalpha is required for tumor necrosis factor inhibition of peroxisome proliferator-activated receptor gamma function. J. Biol. Chem. 2006, 281, 4540–4547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J. Regulation of PPARgamma function by TNF-alpha. Biochem. Biophys. Res. Commun. 2008, 374, 405–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Tian, Z.; Zhuang, X.; Luo, M.; Yin, W.; Xiong, L. The propionic acid and butyric acid in serum but not in feces are increased in patients with diarrhea-predominant irritable bowel syndrome. BMC Gastroenterol. 2020, 20, 73. [Google Scholar] [CrossRef] [Green Version]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-κB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem. 2018, 124, 468–474. [Google Scholar] [CrossRef]
- Azar, S.T.; Tamim, H.; Beyhum, H.N.; Habbal, M.Z.; Almawi, W.Y. Type I (insulin-dependent) diabetes is a Th1- and Th2-mediated autoimmune disease. Clin. Diagn. Lab. Immunol. 1999, 6, 306–310. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J.; Bouzakri, K.; Chevillotte, E.; Ricard, N.; Jacquet, D.; Bastard, J.P.; Laville, M.; Vidal, H. Suppressor of cytokine signaling 3 expression and insulin resistance in skeletal muscle of obese and type 2 diabetic patients. Diabetes 2004, 53, 2232–2241. [Google Scholar] [CrossRef] [Green Version]
- Sireesh, D.; Dhamodharan, U.; Ezhilarasi, K.; Vijay, V.; Ramkumar, K.M. Association of NF-E2 Related Factor 2 (Nrf2) and inflammatory cytokines in recent onset Type 2 Diabetes Mellitus. Sci. Rep. 2018, 8, 5126. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, L.; Qiao, Y.; Zhou, X.; Wu, G.; Wang, L.; Peng, Y.; Dong, X.; Huang, H.; Si, L.; et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE 2013, 8, e75927. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.S.; Lee, M.S. Role of autophagy in diabetes and mitochondria. Ann. N. Y. Acad. Sci. 2010, 1201, 79–83. [Google Scholar] [CrossRef]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Sokolovska, J.; Isajevs, S.; Sugoka, O.; Sharipova, J.; Lauberte, L.; Svirina, D.; Rostoka, E.; Sjakste, T.; Kalvinsh, I.; Sjakste, N. Influence of metformin on GLUT1 gene and protein expression in rat streptozotocin diabetes mellitus model. Arch. Physiol. Biochem. 2010, 116, 137–145. [Google Scholar] [CrossRef]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Xiao, T.; Guan, X.; Nie, L.; Wang, S.; Sun, L.; He, T.; Huang, Y.; Zhang, J.; Yang, K.; Wang, J.; et al. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol. Cell. Biochem. 2014, 394, 145–154. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Rüegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Ogura, Y.; Suzuki, T.; Sen, S.; Lee, S.M.; Kanasaki, K.; Kume, S.; Koya, D. A very-low-protein diet ameliorates advanced diabetic nephropathy through autophagy induction by suppression of the mTORC1 pathway in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Diabetologia 2016, 59, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Towns, R.; Guo, C.; Shangguan, Y.; Hong, S.; Wiley, J.W. Type 2 diabetes with neuropathy: Autoantibody stimulation of autophagy via Fas. Neuroreport 2008, 19, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Tschöp, M.H.; Woods, S.C.; Morton, G.J.; Myers, M.G.; D’Alessio, D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 2013, 503, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouvet, N.; Estall, J.L. The pancreas: Bandmaster of glucose homeostasis. Exp. Cell. Res. 2017, 360, 19–23. [Google Scholar] [CrossRef]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freychet, L.; Rizkalla, S.W.; Desplanque, N.; Basdevant, A.; Zirinis, P.; Tchobroutsky, G.; Slama, G. Effect of intranasal glucagon on blood glucose levels in healthy subjects and hypoglycaemic patients with insulin-dependent diabetes. Lancet 1988, 1, 1364–1366. [Google Scholar] [CrossRef]
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin Signaling in the Control of Glucose and Lipid Homeostasis. Handb. Exp. Pharm. 2016, 233, 51–71. [Google Scholar]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell. Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell. Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Castaño, J.G.; Nieto, A.; Felíu, J.E. Inactivation of phosphofructokinase by glucagon in rat hepatocytes. J. Biol. Chem. 1979, 254, 5576–5579. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, B.B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [Green Version]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Minehira, K. New data and new concepts on the role of the liver in glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [Green Version]
- Adeva-Andany, M.M.; Pérez-Felpete, N.; Fernández-Fernández, C.; Donapetry-García, C.; Pazos-García, C. Liver glucose metabolism in humans. Biosci. Rep. 2016, 36, e00416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moszak, M.; Szulińska, M.; Bogdański, P. You Are What You Eat-The Relationship between Diet, Microbiota, and Metabolic Disorders-A Review. Nutrients 2020, 12, 1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvais-Jarvis, F. Gender differences in glucose homeostasis and diabetes. Physiol. Behav. 2018, 187, 20–23. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Jena, G. The role of butyrate, a histone deacetylase inhibitor in diabetes mellitus: Experimental evidence for therapeutic intervention. Epigenomics 2015, 7, 669–680. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, J.; Zhang, Y.; Tang, J.; Sun, B.; Xu, W.; Wang, X.; Chen, Y.; Sun, Z. Changes in Intestinal Microbiota Are Associated with Islet Function in a Mouse Model of Dietary Vitamin A Deficiency. J. Diabetes Res. 2020, 2020, 2354108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling—Mechanisms and research needs. Nat. Rev. Endocrinol. 2019, 15, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wei, J.; Liu, P.; Zhang, Q.; Tian, Y.; Hou, G.; Meng, L.; Xin, Y.; Jiang, X. Role of the gut microbiota in type 2 diabetes and related diseases. Metabolism 2021, 117, 154712. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Okar, D.A.; Kang, J.; Lange, A.J. Reduction of hepatic glucose production as a therapeutic target in the treatment of diabetes. Curr. Drug. Targets. Immune. Endocr. Metabol. Disord. 2005, 5, 51–59. [Google Scholar] [CrossRef]
- Lu, P.; Chen, X.; Zhang, Z.; Zhang, J.; Yang, Y.; Liu, Z.; Xie, J.; Shao, S.; Zhou, X.; Hu, S.; et al. Insulin upregulates betatrophin expression via PI3K/Akt pathway. Sci. Rep. 2017, 7, 5594. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Doss, C.G.; Bandyopadhyay, S.; Agoramoorthy, G. Influence of miRNA in insulin signaling pathway and insulin resistance: Micro-molecules with a major role in type-2 diabetes. Wiley. Interdiscip. Revs. RNA 2014, 5, 697–712. [Google Scholar] [CrossRef]
- Tan, C.Y.; Weier, Q.; Zhang, Y.; Cox, A.J.; Kelly, D.J.; Langham, R.G. Thioredoxin-interacting protein: A potential therapeutic target for treatment of progressive fibrosis in diabetic nephropathy. Nephron 2015, 129, 109–127. [Google Scholar] [CrossRef]
- Abulizi, A.; Cardone, R.L.; Stark, R.; Lewandowski, S.L.; Zhao, X.; Hillion, J.; Ma, L.; Sehgal, R.; Alves, T.C.; Thomas, C.; et al. Multi-Tissue Acceleration of the Mitochondrial Phosphoenolpyruvate Cycle Improves Whole-Body Metabolic Health. Cell. Metab. 2020, 32, 751–766. [Google Scholar] [CrossRef]
- Lewandowski, S.L.; Cardone, R.L.; Foster, H.R.; Ho, T.; Potapenko, E.; Poudel, C.; VanDeusen, H.R.; Sdao, S.M.; Alves, T.C.; Zhao, X.; et al. Pyruvate Kinase Controls Signal Strength in the Insulin Secretory Pathway. Cell. Metab. 2020, 32, 736–750. [Google Scholar] [CrossRef]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, S.; Liaskou, E.; Fear, J.; Garg, A.; Reynolds, G.; Claridge, L.; Adams, D.H.; Newsome, P.N.; Lalor, P.F. Dysregulated hepatic expression of glucose transporters in chronic disease: Contribution of semicarbazide-sensitive amine oxidase to hepatic glucose uptake. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G1180–G1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflug. Archiv. 2020, 472, 1273–1298. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukkanen, O.; Lindström, J. Finnish Diabetes Prevention Study: Polymorphisms in the SLC2A2 (GLUT2) gene are associated with the conversion from impaired glucose tolerance to type 2 diabetes: The Finnish Diabetes Prevention Study. Diabetes 2005, 54, 2256–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burcelin, R.; del Carmen Muñoz, M.; Guillam, M.T.; Thorens, B. Liver hyperplasia and paradoxical regulation of glycogen metabolism and glucose-sensitive gene expression in GLUT2-null hepatocytes. Further evidence for the existence of a membrane-based glucose release pathway. J. Biol. Chem. 2000, 275, 10930–10936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enogieru, O.J.; Ung, P.M.U.; Yee, S.W.; Schlessinger, A.; Giacomini, K.M. Functional and structural analysis of rare SLC2A2 variants associated with Fanconi-Bickel syndrome and metabolic traits. Human 2019, 40, 983–995. [Google Scholar]
- Gorovits, N.; Cui, L.; Busik, J.V.; Ranalletta, M.; Hauguel de-Mouzon, S.; Charron, M.J. Regulation of hepatic GLUT8 expression in normal and diabetic models. Endocrinology 2003, 144, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- Keembiyehetty, C.; Augustin, R.; Carayannopoulos, M.O.; Steer, S.; Manolescu, A.; Cheeseman, C.I.; Moley, K.H. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are up-regulated in diabetes. Mol. Endocrinol. 2006, 20, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Tal, M.; Kahn, B.B.; Lodish, H.F. Expression of the low Km GLUT-1 glucose transporter is turned on in perivenous hepatocytes of insulin-deficient diabetic rats. Endocrinology 1991, 129, 1933–1941. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in the small intestine in health and disease. Pflug. Arch. 2020, 472, 1207–1248. [Google Scholar] [CrossRef] [PubMed]
- Charrez, B.; Qiao, L.; Hebbard, L. The role of fructose in metabolism and cancer. Horm. Mol. Biol. Clin. Investig. 2015, 22, 79–89. [Google Scholar] [CrossRef]
- Karim, S.; Adams, D.H.; Lalor, P.F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 2012, 18, 6771–6781. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prats, C.; Graham, T.E.; Shearer, J. The dynamic life of the glycogen granule. J. Biol. Chem. 2018, 293, 7089–7098. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef]
- Jensen, T.E.; Richter, E.A. Regulation of glucose and glycogen metabolism during and after exercise. J. Physiol. 2012, 590, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Xirouchaki, C.E.; Mangiafico, S.P.; Bate, K.; Ruan, Z.; Huang, A.M.; Tedjosiswoyo, B.W.; Lamont, B.; Pong, W.; Favaloro, J.; Blair, A.R.; et al. Impaired glucose metabolism and exercise capacity with muscle-specific glycogen synthase 1 (gys1) deletion in adult mice. Mol. Metab. 2016, 5, 221–232. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen metabolism in humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Katz, A. A century of exercise physiology: Key concepts in regulation of glycogen metabolism in skeletal muscle. Eur. J. Appl. Physiol. 2022, 122, 1751–1772. [Google Scholar] [CrossRef]
- Härndahl, L.; Schmoll, D.; Herling, A.W.; Agius, L. The role of glucose 6-phosphate in mediating the effects of glucokinas e overexpression on hepatic glucose metabolism. FEBS J. 2006, 273, 336–346. [Google Scholar] [CrossRef]
- Jensen, J.; Lai, Y.C. Regulation of muscle glycogen synthase phosphorylation and kinetic properties by insulin, exercise, adrenaline and role in insulin resistance. Arch. Physiol. Biochem. 2009, 115, 13–21. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E413–E422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, J.; Di, L.J. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, L. Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J. Mol. Endocrinol. 2016, 57, R93–R108. [Google Scholar] [CrossRef] [Green Version]
- Sternisha, S.M.; Miller, B.G. Molecular and cellular regulation of human glucokinase. Arch. Biochem. Biophys. 2019, 663, 199–213. [Google Scholar] [CrossRef]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset d iabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.I.; Polonsky, K.S. Diabetes mellitus and genetically programmed defects in beta-cell func tion. Nature 2001, 414, 788–791. [Google Scholar] [CrossRef]
- Kim, S.H. Maturity-Onset Diabetes of the Young: What Do Clinicians Need to Know? Diabetes Metab. J. 2015, 39, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Fernandes Silva, L.; Vangipurapu, J.; Kuulasmaa, T.; Laakso, M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci. Rep. 2019, 9, 10240. [Google Scholar] [CrossRef] [Green Version]
- Degerman, E.; Ahmad, F.; Chung, Y.W.; Guirguis, E.; Omar, B.; Stenson, L.; Manganiello, V. From PDE3B to the regulation of energy homeostasis. Curr. Opin. Pharmacol. 2011, 11, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Tian, X.; Wu, H.; Huang, J.; Li, M.; Mei, Z.; Zhou, L.; Xie, H.; Zheng, S. Metabolic Changes of Hepatocytes in NAFLD. Front. Physiol. 2021, 12, 710420. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, C. Gluconeogenesis in Cancer: Function and Regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 2019, 5, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef]
- Jitrapakdee, S. Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell. Biol. 2012, 44, 33–45. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Ding, X.; Wang, Y.; Gu, M.; Zhang, J.; Yan, S.; Li, N.; Song, Z.; Yin, J.; Lu, L.; et al. Spexin alleviates insulin resistance and inhibits hepatic gluconeogenesis via the FoxO1/PGC-1α pathway in high-fat-diet-induced rats and insulin resistant cells. Int. J. Biol. Sci. 2019, 15, 2815–2829. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [Green Version]
- Rahnert, J.A.; Zheng, B.; Hudson, M.B.; Woodworth-Hobbs, M.E.; Price, S.R. Glucocorticoids Alter CRTC-CREB Signaling in Muscle Cells: Impact on PGC-1α Expression and Atrophy Markers. PLoS ONE 2016, 11, e0159181. [Google Scholar] [CrossRef] [Green Version]
- Besseiche, A.; Riveline, J.P.; Gautier, J.F.; Bréant, B.; Blondeau, B. Metabolic roles of PGC-1α and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Jiang, A.; Takizawa, F.; Safdar, A.; Manika, A.; Tesmenitsky, Y.; Kang, K.T.; Bischoff, J.; Kalwa, H.; Sartoretto, J.L.; et al. Endothelial PGC-1α mediates vascular dysfunction in diabetes. Cell. Metab. 2014, 19, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Zhang, Y.; Li, J.; Hou, D.; Xiang, Y. Effect of PGC-1α on proliferation, migration, and transdifferentiation of rat vascular smooth muscle cells induced by high glucose. J. Biomed. Biotechnol. 2012, 2012, 756426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rullman, E.; Fernandez-Gonzalo, R.; Mekjavić, I.B.; Gustafsson, T.; Eiken, O. MEF2 as upstream regulator of the transcriptome signature in human skeletal muscle during unloading. Am. J. Physiol. Regul. Integl. Comp. Physiol. 2018, 315, R799–R809. [Google Scholar] [CrossRef] [Green Version]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte Nuclear Factor 4α (Nuclear Receptor 2A1) Is Essential for M aintenance of Hepatic Gene Expression and Lipid Homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Babeu, J.P.; Boudreau, F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J. Gastroenterol. 2014, 20, 22–30. [Google Scholar] [CrossRef]
- Duncan, S.A.; Manova, K.; Chen, W.S.; Hoodless, P.; Weinstein, D.C.; Bachvarova, R.F.; Darnell, J.E. Expression of transcription factor HNF-4 in the extraembryonic endoder m, gut, and nephrogenic tissue of the developing mouse embryo: HNF-4 is a marker for primary endoderm in the implanting blastocyst. Proc. Natl. Acad. Sci. USA 1994, 91, 7598–7602. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Xie, Z.; Cao, D.; Gong, M.; Yang, L.; Zhou, Z.; Ou, Y. C-Phycocyanin inhibits hepatic gluconeogenesis and increases glycogen synthesis via activating Akt and AMPK in insulin resistance hepatocytes. Food. Func. 2018, 9, 2829–2839. [Google Scholar] [CrossRef]
- Yamagata, K.; Furuta, H.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Cox, N.J.; Fajans, S.S.; Signorini, S.; Stoffel, M.; Bell, G.I. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-ons et diabetes of the young (MODY1). Nature 1996, 384, 458–460. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Ong, R.T.H.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat. Genet. 2011, 44, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.; Raeder, H.; Eide, S.A.; Midthjell, K.; Hveem, K.; Søvik, O.; Molven, A.; Njølstad, P.R. Studies in 3,523 Norwegians and meta-analysis in 11,571 subjects indic ate that variants in the hepatocyte nuclear factor 4 alpha (HNF4A) P2 region are associated with type 2 diabetes in Scandinavians. Diabetes 2007, 56, 3112–3117. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Maechler, P.; Antinozzi, P.A.; Hagenfeldt, K.A.; Wollheim, C.B. Hepatocyte nuclear factor 4alpha regulates the expression of pancreati c beta-cell genes implicated in glucose metabolism and nutrient-induc ed insulin secretion. J. Biol. Chem. 2000, 275, 35953–35959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metab olism. Nature 2005, 437, 1109–1111. [Google Scholar] [CrossRef]
- Wei, S.; Zhang, M.; Yu, Y.; Xue, H.; Lan, X.; Liu, S.; Hatch, G.; Chen, L. HNF-4α regulated miR-122 contributes to development of gluconeogenesis and lipid metabolism disorders in Type 2 diabetic mice and in palmitate-treated HepG2 cells. Eur. J. Pharmacol. 2016, 791, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Shi, X.; Zhu, J.; Guan, X.; Garbacz, W.G.; Huang, Y.; Gao, L.; Yan, J.; Xu, M.; Ren, S.; et al. Regulation of Cholesterol Sulfotransferase SULT2B1b by Hepatocyte Nucl ear Factor 4α Constitutes a Negative Feedback Control of Hepatic Gluco neogenesis. Mol. Cell. Biol. 2018, 38, e00654-17. [Google Scholar] [CrossRef] [Green Version]
- Calissi, G.; Lam, E.W.; Link, W. Therapeutic strategies targeting FOXO transcription factors. Nat. Rev. Drug Discov. 2021, 20, 21–38. [Google Scholar] [CrossRef]
- Kim, H.N.; Iyer, S.; Ring, R.; Almeida, M. The Role of FoxOs in Bone Health and Disease. Curr. Top. Dev. Biol. 2018, 127, 149–163. [Google Scholar] [PubMed]
- Liu, T.Y.; Shi, C.X.; Gao, R.; Sun, H.J.; Xiong, X.Q.; Ding, L.; Chen, Q.; Li, Y.H.; Wang, J.J.; Kang, Y.M.; et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin. Sci. 2015, 129, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; White, M.F. Targeting Forkhead box O1 from the concept to metabolic diseases: Lessons from mouse models. Antioxid. Redox Signal. 2011, 14, 649–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Wang, Y.; Liang, W.; Tang, X.; Zhang, Y.; Wang, L.; Zhao, P.; Lu, Z. Bombyx mori nucleopolyhedrovirus downregulates transcription factor BmFoxO to elevate virus infection. Dev. Comp. Immunol. 2021, 116, 103904. [Google Scholar] [CrossRef]
- Kitamura, T. The role of FOXO1 in β-cell failure and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 615–623. [Google Scholar] [CrossRef]
- Nakae, J.; Biggs, W.H., 3rd; Kitamura, T.; Cavenee, W.K.; Wright, C.V.; Arden, K.C.; Accili, D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat. Genet. 2002, 32, 245–253. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, X.; Yan, H.; Wu, Y.; Pan, Q.; Shen, J.Z.; Li, X.; Chen, Y.; Li, L.; Qi, Y.; et al. Phosphorylation of Forkhead Protein FoxO1 at S253 Regulates Glucose Homeostasis in Mice. Endocrinology 2019, 160, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Li, J.; Feng, S.; Li, Y.; Tan, L. Long noncoding RNA Gomafu upregulates Foxo1 expression to promote hepatic insulin resistance by sponging miR-139-5p. Cell Death. Dis. 2018, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Langlet, F.; Haeusler, R.A.; Lindén, D.; Ericson, E.; Norris, T.; Johansson, A.; Cook, J.R.; Aizawa, K.; Wang, L.; Buettner, C.; et al. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 2017, 171, 824–835. [Google Scholar] [CrossRef]
- Gombos, Z.; Koltai, E.; Torma, F.; Bakonyi, P.; Kolonics, A.; Aczel, D.; Ditroi, T.; Nagy, P.; Kawamura, T.; Radak, Z. Hypertrophy of Rat Skeletal Muscle Is Associated with Increased SIRT1/Akt/mTOR/S6 and Suppressed Sestrin2/SIRT3/FOXO1 Levels. Int. J. Mol. Sci. 2021, 22, 7588. [Google Scholar] [CrossRef]
- Gopal, K.; Saleme, B.; Al Batran, R.; Aburasayn, H.; Eshreif, A.; Ho, K.L.; Ma, W.K.; Almutairi, M.; Eaton, F.; Gandhi, M.; et al. FoxO1 regulates myocardial glucose oxidation rates via transcriptional control of pyruvate dehydrogenase kinase 4 expression. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H479–H490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Perdomo, G.; Zhang, T.; Slusher, S.; Lee, S.; Phillips, B.E.; Fan, Y.; Giannoukakis, N.; Gramignoli, R.; Strom, S.; et al. FoxO6 integrates insulin signaling with gluconeogenesis in the liver. Diabetes 2011, 60, 2763–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scapin, G.; Dandey, V.P.; Zhang, Z.; Prosise, W.; Hruza, A.; Kelly, T.; Mayhood, T.; Strickland, C.; Potter, C.S.; Carragher, B. Structure of the insulin receptor–insulin complex by single-particle cryo-EM analysis. Nature 2021, 556, 122–125. [Google Scholar] [CrossRef]
- Kahn, C.R.; Flier, J.S.; Bar, R.S.; Archer, J.A.; Gorden, P.; Martin, M.M.; Roth, J. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N. Engl. J. Med. 1976, 294, 739–745. [Google Scholar] [CrossRef]
- Taylor, S.I. Lilly Lecture: Molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin-receptor gene. Diabetes 1992, 41, 1473–1490. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Copps, K.D.; Dong, X.; Park, S.; Cheng, Z.; Pocai, A.; Rossetti, L.; Sajan, M.; Farese, R.V.; White, M.F. The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol. Cell. Biol. 2009, 29, 5070–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Wei, T. Inputs and outputs of insulin receptor. Protein Cell 2014, 5, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, A.; White, M.F. Insulin-like signaling, nutrient homeostasis, and life span. Annu. Rev. Physiol. 2008, 70, 191–212. [Google Scholar] [CrossRef]
- Poretsky, L.; Cataldo, N.A.; Rosenwaks, Z.; Giudice, L.C. The insulin-related ovarian regulatory system in health and disease. Endocr. Rev. 1999, 20, 535–582. [Google Scholar] [CrossRef]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell. Biochem. 1998, 182, 31–48. [Google Scholar] [CrossRef]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 1999, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.H.; Sarnecki, C.; Blenis, J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 1998, 12, 915–927. [Google Scholar]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2012, 81, 807–869. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, U.S.; Ram, P.T.; Iyengar, R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 2002, 297, 1018–1023. [Google Scholar] [CrossRef] [Green Version]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1β-Induced Insulin Resistance in Adipocytes through Down-R egulation of Insulin Receptor Substrate-1 Expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Thomson, J.K.; Zhao, W.; Wu, X.; Gao, X.; DeMarco, D.; Kong, W.; Tong, M.; Sun, J.; Bakhos, M.; et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J. Mol. Cell. Cardiol. 2018, 114, 105–115. [Google Scholar] [CrossRef]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomai ns, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A Stress Signaling Pathway in Adipose Tissue Regulates Hepatic Insulin Resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Aouadi, M.; Caron, L.; Even, P.; Belmonte, N.; Prot, M.; Dani, C.; Hofman, P.; Pagès, G.; Pouysségur, J.; et al. The Extracellular Signal-Regulated Kinase Isoform ERK1 Is Specifically Required for In Vitro and In Vivo Adipogenesis. Diabetes 2005, 54, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreelli, F.; Laville, M.; Ducluzeau, P.H.; Vega, N.; Vallier, P.; Khalfallah, Y.; Riou, J.P.; Vidal, H. Defective regulation of phosphatidylinositol-3-kinase gene expression in skeletal muscle and adipose tissue of non-insulin-dependent diabetes mellitus patients. Diabetologia 1999, 42, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadowaki, T. Insights into insulin resistance and type 2 diabetes from knockout mouse models. J. Clin. Investig. 2000, 106, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.Y.; Zhou, Q.L.; Coleman, K.A.; Chouinard, M.; Boese, Q.; Czech, M.P. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 7569–7574. [Google Scholar] [CrossRef] [Green Version]
- Buzzi, F.; Xu, L.; Zuellig, R.A.; Boller, S.B.; Spinas, G.A.; Hynx, D.; Chang, Z.; Yang, Z.; Hemmings, B.A.; Tschopp, O.; et al. Differential effects of protein kinase B/Akt isoforms on glucose homeostasis and islet mass. Mol. Cell. Biol. 2010, 30, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B., 3rd; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacki ng the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Mora, A.; Komander, D.; van Aalten, D.M.F.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell. Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, A.; Lipina, C.; Tronche, F.; Sutherland, C.; Alessi, D.R. Deficiency of PDK1 in liver results in glucose intolerance, impairment of insulin-regulated gene expression and liver failure. Biochem. J. 2005, 385, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Kido, Y.; Uchida, T.; Asahara, S.; Shigeyama, Y.; Matsuda, T.; Takeda, A.; Tsuchihashi, D.; Nishizawa, A.; Ogawa, W.; et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat. Genet. 2006, 38, 589–593. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug. Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, N.; Kesharwani, D.; Datta, M. Lnc-ing non-coding RNAs with metabolism and diabetes: Roles of lncRNAs. Cell. Mol. Life Sci. 2018, 75, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Tan, Q.; Ma, J.; Zhang, J.; Yu, P. Emerging Role of LncRNA Regulation for NLRP3 Inflammasome in Diabetes Complications. Front. Cell Dev. Biol. 2022, 9, 792401. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, L.; Esguerra, J.L. Role of non-coding RNAs in pancreatic beta-cell development and physiology. Acta Physiol. 2014, 211, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death. Dis. 2014, 5, e1506. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wu, Y.B.; Zhou, J.; Kang, D.M. Upregulation of lncRNA MEG3 promotes hepatic insulin resistance via increasing FoxO1 expression. Biochem. Biophys. Res. Commun. 2016, 469, 319–325. [Google Scholar] [CrossRef]
- Ruan, X.; Li, P.; Cangelosi, A.; Yang, L.; Cao, H. A Long Non-coding RNA, lncLGR, Regulates Hepatic Glucokinase Expression and Glycogen Storage during Fasting. Cell. Rep. 2016, 14, 1867–1875. [Google Scholar] [CrossRef] [Green Version]
- Goyal, N.; Sivadas, A.; Shamsudheen, K.V.; Jayarajan, R.; Verma, A.; Sivasubbu, S.; Scaria, V.; Datta, M. RNA sequencing of db/db mice liver identifies lncRNA H19 as a key regulator of gluconeogenesis and hepatic glucose output. Sci. Rep. 2017, 7, 8312. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Xu, R.; Gerin, I.; Cawthorn, W.P.; Macdougald, O.A.; Chen, X.W.; Saltiel, A.R.; Koenig, R.J.; Xu, B. SRA regulates adipogenesis by modulating p38/JNK phosphorylation and stimulating insulin receptor gene expression and downstream signaling. PLoS ONE 2014, 9, e95416. [Google Scholar] [CrossRef] [Green Version]
- Tennant, B.R.; Robertson, A.G.; Kramer, M.; Li, L.; Zhang, X.; Beach, M.; Thiessen, N.; Chiu, R.; Mungall, K.; Whiting, C.J.; et al. Identification and analysis of murine pancreatic islet enhancers. Diabetologia 2013, 56, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.L.; Wang, F.F.; Shu, J.; Tian, S.; Jiang, Y.; Zhang, D.; Wang, N.; Luo, Q.; Zhang, Y.; Jin, F.; et al. Transgenerational glucose intolerance with Igf2/H19 epigenetic alterations in mouse islet induced by intrauterine hyperglycemia. Diabetes 2012, 61, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, D.D.; Zhang, E.B.; You, L.H.; Wang, N.; Wang, L.T.; Jin, F.Y.; Zhu, Y.N.; Cao, L.H.; Yuan, Q.X.; De, W.; et al. Downregulation of lncRNA TUG1 affects apoptosis and insulin secretion in mouse pancreatic β cells. Cell. Physiol. Biochem. 2015, 35, 1892–1904. [Google Scholar] [CrossRef]
- Yau, J.W.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Meta-Analysis for Eye Disease (META-EYE) Study Group. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Yan, B.; Tao, Z.F.; Li, X.M.; Zhang, H.; Yao, J.; Jiang, Q. Aberrant expression of long noncoding RNAs in early diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 941–951. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Dan, Y.; Gao, X.; Huang, L.; Lv, H.; Chen, J. DNMT1-mediated lncRNA MEG3 methylation accelerates endothelial-mesenchymal transition in diabetic retinopathy through the PI3K/Akt/mTOR signaling pathway. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E598–E608. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.; Yafai, Y.; Noack, A.; Yang, X.M.; Munk, A.B.; Krohn, S.; Iandiev, I.; Wiedemann, P.; Reichenbach, A.; Eichler, W. The axon guidance molecule Netrin-4 is expressed by Müller cells and contributes to angiogenesis in the retina. Glia 2012, 60, 1567–1578. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Kowluru, R.A. Long Noncoding RNA MALAT1 and Regulation of the Antioxidant Defense System in Diabetic Retinopathy. Diabetes 2021, 70, 227–239. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Tian, W.; Fu, H.T.; Li, C.P.; Liu, B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef]
- Feng, S.D.; Yang, J.H.; Yao, C.H.; Yang, S.S.; Zhu, Z.M.; Wu, D.; Ling, H.Y.; Zhang, L. Potential regulatory mechanisms of lncRNA in diabetes and its complications. Biochem. Cell Biol. 2017, 95, 361–367. [Google Scholar] [CrossRef]
- Alvarez, M.L.; DiStefano, J.K. Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS ONE 2011, 6, e18671. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.L.; Khosroheidari, M.; Eddy, E.; Kiefer, J. Role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: Implications for diabetic nephropathy. PLoS ONE 2013, 8, e77468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, H.; Peng, R.; Zhang, L.Y.; Sun, Y.; Peng, H.M.; Liu, H.D.; Yu, L.J.; Li, A.L.; Zhang, Y.J.; Jiang, W.H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death. Dis. 2017, 8, e2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Wang, M.; Chen, Z.; Bhatt, K.; Oh, H.J.; Lanting, L.; Deshpande, S.; Jia, Y.; Lai, J.Y.; O’Connor, C.L.; et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Commun. 2016, 7, 12864. [Google Scholar] [CrossRef] [Green Version]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Gu, H.; Xu, W.; Zhou, X. Down-regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int. J. Cardiol. 2016, 203, 214–216. [Google Scholar] [CrossRef]
- Zhang, M.; Gu, H.; Chen, J.; Zhou, X. Involvement of long noncoding RNA MALAT1 in the pathogenesis of diabetic cardiomyopathy. Int. J. Cardiol. 2016, 202, 753–755. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Yao, B.; Xu, W.; Chen, J.; Zhou, X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci. Rep. 2016, 6, 36340. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Huda, M.N.; Kim, M.; Bennett, B.J. Modulating the Microbiota as a Therapeutic Intervention for Type 2 Diabetes. Front. Endocrinol. 2021, 12, 632335. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, H.; Zhan, L.; Lu, X.; Zhang, L. Dynamic Development of Fecal Microbiome During the Progression of Diabetes Mellitus in Zucker Diabetic Fatty Rats. Front. Microbiol. 2019, 10, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Jia, Q.; Mehmood, S.; Ma, S.; Liu, X. Genistein ameliorates inflammation and insulin resistance through mediation of gut microbiota composition in type 2 diabetic mice. Eur. J. Nutr. 2021, 60, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Matheus, V.A.; Monteiro, L.; Oliveira, R.B.; Maschio, D.A.; Collares-Buzato, C.B. Butyrate reduces high-fat diet-induced metabolic alterations, hepatic steatosis and pancreatic beta cell and intestinal barrier dysfunctions in prediabetic mice. Exp. Biol. Med. 2017, 242, 1214–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Sun, B.; Yu, D.; Zhu, C. Gut Microbiota: An Important Player in Type 2 Diabetes Mellitus. Front. Cell Infect. Microbiol. 2022, 12, 834485. [Google Scholar] [CrossRef]
- Fröjdö, S.; Durand, C.; Molin, L.; Carey, A.L.; El-Osta, A.; Kingwell, B.A.; Febbraio, M.A.; Solari, F.; Vidal, H.; Pirola, L. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol. Cell. Endocrinol. 2011, 335, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Song, M.Y.; Song, E.K.; Kim, E.K.; Moon, W.S.; Han, M.K.; Park, J.W.; Kwon, K.B.; Park, B.H. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 2009, 58, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Koya, D. SIRT1 in Type 2 Diabetes: Mechanisms and Therapeutic Potential. Diabetes Metab. J. 2013, 37, 315–325. [Google Scholar] [CrossRef]
- Lemos, V.; de Oliveira, R.M.; Naia, L.; Szegö, É.; Ramos, E.; Pinho, S.; Magro, F.; Cavadas, C.; Rego, A.C.; Costa, V.; et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 2017, 26, 4105–4117. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Wang, S.; Xiao, M.; Lin, Y.; Zhou, L.; Lei, Q.; Xiong, Y.; Guan, K.L.; Zhao, S. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol. Cell 2011, 43, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Dong, X.; Wang, X.; Zheng, Y.; Qiu, J.; Peng, Y.; Xu, J.; Chai, Z.; Liu, C. Multiple Roles of SIRT2 in Regulating Physiological and Pathological Signal Transduction. Genet. Res. 2022, 2022, 9282484. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Transcription Factors | Related Processes | Related Genes/Proteins |
|---|---|---|
| PGC-1α | Hepatic insulin resistance; fatty acid oxidation | PPARα |
| Hepatic gluconeogenesis | CREB, CRTC2 | |
| Insulin secretion | AMPK signal pathway | |
| The uptake of glucose | MEF2 | |
| Glycogen synthesis | PDK4 | |
| HNF4α | Gluconeogenesis | PGC-1α; CS; SULT2B1b |
| Gluconeogenesis; lipid metabolism | DR-1 MiR-122 | |
| Foxo | Gluconeogenesis | G6Pase; PEPCK; PGC-1α; PI3K/AKT |
| Gluconeogenesis | LncRNA Gomafu; miR-139 | |
| Glucose consumption | PDK4 | |
| Glucose production | Sin3a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Wu, H.; Li, Z. The Pathogenesis of Diabetes. Int. J. Mol. Sci. 2023, 24, 6978. https://doi.org/10.3390/ijms24086978
Guo H, Wu H, Li Z. The Pathogenesis of Diabetes. International Journal of Molecular Sciences. 2023; 24(8):6978. https://doi.org/10.3390/ijms24086978
Chicago/Turabian StyleGuo, Huiqin, Haili Wu, and Zhuoyu Li. 2023. "The Pathogenesis of Diabetes" International Journal of Molecular Sciences 24, no. 8: 6978. https://doi.org/10.3390/ijms24086978