In this section, we present the proposed BioGraph data model, the methods we used to implement the model and the user-friendly interface, and the materials used to validate the implemented model.
3.1. Model Definition and Implementation
We have defined a set of goals that the ideal data model should fulfill so the integrated search of metadata sourced from various datasets could be achieved. Those goals are:
Unified representation of metadata from diverse formats and schemas.
Efficient querying and finding complex patterns in data.
Allowing easy extensibility through schema extensions without affecting the core data model.
Being database-agnostic as much as possible, so the underlying database management system and related software do not dictate the model’s design.
Biological data contain information about biological entities and their properties. We recognized those entities as the primary building blocks of the new model. Entities are not isolated objects but are highly interconnected in many ways. The relations between the entities have various origins, from biological processes to taxonomical relations and mutual similarities. Associating entities from multiple datasets are enabled by connecting relations paths between them across multiple datasets. As many types of entities exist, creating specific model schemas for each biological entity and their relations is inefficient. It violates the requirement of unified representation, and there is no feasible way of predicting all future properties and relations associated with every entity type. That is why we decided to maintain a model which enables storing metadata of arbitrary entity types in a generic form. Details regarding entity type-specific information are delegated to high-level data schemas. It is essential to mention that our model links metadata related to biological entities in a way that can be used for efficient data location and retrieval from the original data sources. An example of gene metadata might be its identifier, while the data are a DNA sequence representing the gene. The gene sequence is not handled by the model but can be easily retrieved from the external database, such as the NCBI gene, using the metadata linked in the model.
3.2. Model Definition
Our model consists of three object types: entity objects, identifier objects, data objects, and relations that connect the objects. Model objects and their individual relationships are shown in
Figure 1. Metadata are loaded from arbitrary data formats, from which entities, identifiers, and data objects are extracted along with respective relations. The extracted objects and relations form a unified knowledge graph ready for searching, having each object represented as a graph node connected to other nodes using the relation edges.
Details about all created objects and relations are stored in a catalog database for additional indexing.
Figure 2 shows a small segment of the constructed knowledge graph, displayed using the Neo4J browser.
Biological entities come in different types. Some are associated with biological functions, such as genes or proteins, while others can be more general, such as habitats and coverage areas. Regardless of the type, each entity can be assigned at least one unique identifier, which we denote as a primary identifier. When there are multiple unique identifiers, the primary identifier is chosen to be the one shared between most biological datasets. An example of a primary identifier for a gene entity is a gene name. It is important to note that the data model is not restricted to human genetic data, but can also support data from arbitrary biological domains regardless of the data source. We use entity type names and primary identifiers to construct entity objects in our data model. Each entity object represents a single biological entity.
Figure 3 shows concrete examples of biological objects (sourced from DisProt [
19], DisGeNET [
20], Tantigen 2.0 [
21], IEDB [
22], and HGNC [
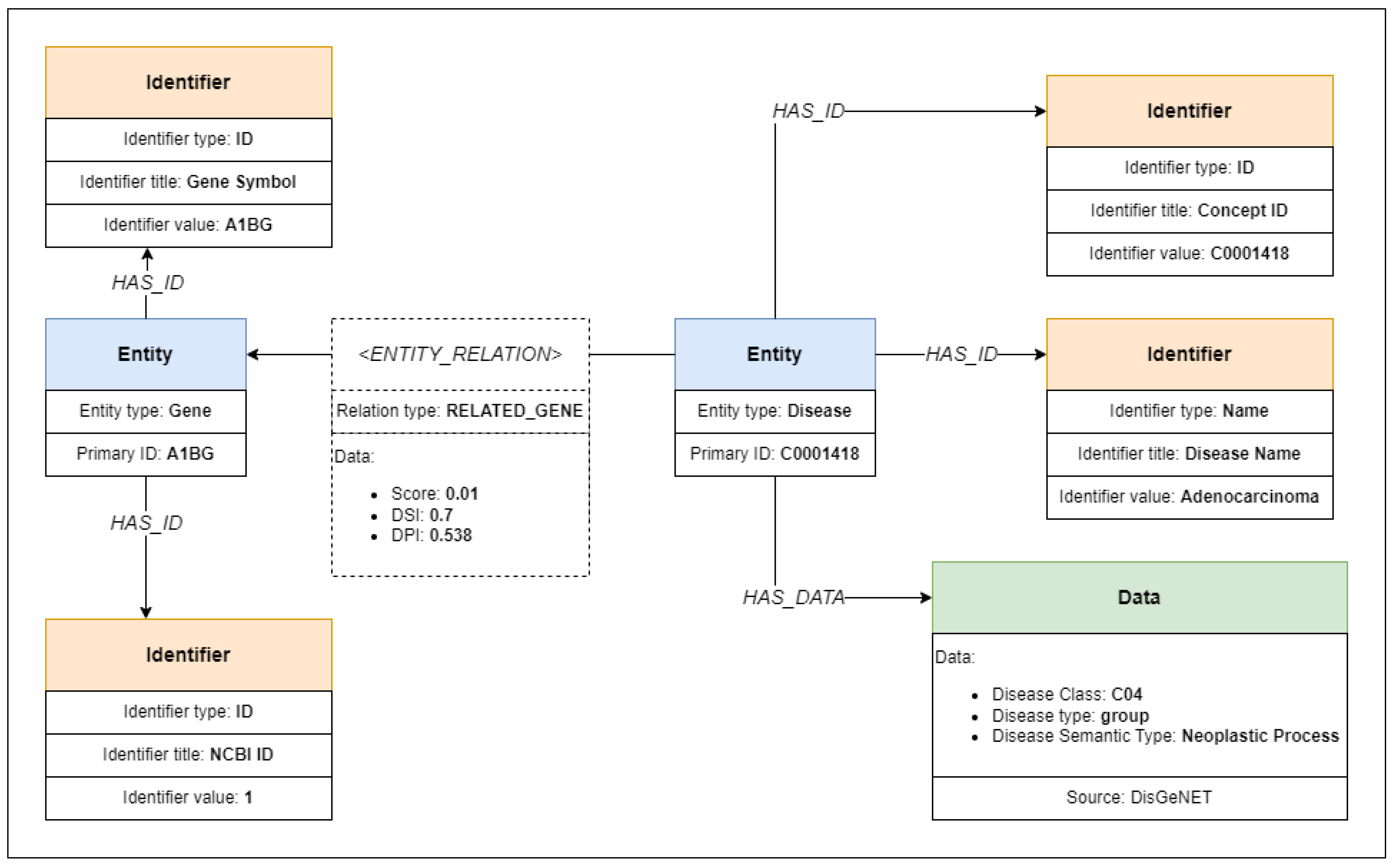
23] datasets) and their relations. Five different entities are in blue boxes along with their type label and primary identifiers. Those entities, with their respective primary identifiers, are gene CDKN1A, protein P38936, disease C0038356, antigen Ag002102, and epitope with the sequence FAWERVRGL.
All available entity identifiers, including the primary identifier, are collected and represented in our model using identifier objects. The identifier objects are connected to their respective entity objects using identifier relations labeled as “HAS ID” relations. Each identifier contains information about the identifier type, title, and value. The identifier type defines the nature of the identifier—a name, a URL, or a generic identifier without any specific type. The identifier title is used to determine the meaning of the identifier, such as “gene name” or “NCBI ID”. Finally, the value stores the string value of the identifier. One identifier can be shared between multiple entities, so one identifier object can be connected to multiple entity objects. Besides identifiers, datasets may contain additional metadata about the entities, such as protein regions, location coordinates, or gene positions on chromosomes. We collect those metadata values and store them in data objects, along with the source label so that the metadata can be easily tracked and verified against the original dataset.
Figure 3 show six identifiers in yellow boxes, connected to five entities. The example protein entity is connected with two identifiers, where P38936 is the identifier of the protein in the UniProt [
24] database (also used as a primary identifier) and DP00016 is the identifier of the same protein in DisProt [
19] database.
The data objects, containing the metadata of the entities, that are not identifiers, are associated with their respective entities using data relations labeled as “HAS DATA” relations. The example in
Figure 3 shows data objects that give additional information to entities. A data object connected to a protein entity contains disorder content value, gene metadata contain gene location on chromosome, disease metadata contain disease type annotation, antigen metadata contain the full name of the antigen, and epitope data contain type annotation for the epitope.
Relations can also exist between entities. A protein can be associated with its source gene, and a blood sample can be associated with the laboratory where it was collected. As the entity relations are various, they are defined in high-level schemas. Base relations between entities are displayed in
Table 2, but the list can be extended using higher-level schemas. All relations in our model share the same structure, containing information about the relation label and optional data specifying the details of a connection between the associated entities.
An example network of biological objects from five different sources (DisProt [
19], DisGeNET [
20], Tantigen [
21], IEDB [
22], and HGNC [
23]) represented using the BioGraph model elements is displayed in
Figure 3.
Duplicate entries can negatively affect search results and the overall performance of searching. Our model assigns specific identifiers to objects and relations based on their content to prevent duplicate entries. The method for generating such identifiers is called content addressing. Data stored in an object or relation are mapped to a number from a large interval using a hash function, with a very low probability of having multiple different data mapped to the same number. If the mapping is performed multiple times on the same object, it will always result in the same number, and the resulting number can be used as a unique identifier.
3.3. Model Implementation
We have tested multiple database systems for storing metadata objects. Although the model was successfully mapped to relation databases, querying speeds were not satisfying, so we decided to use a graph database system as the underlying database. The Neo4J [
26] graph database system provided the best overall results, but the model is not restricted to a specific data storage system. We used a relational database as a catalog database to store the import metadata of all objects and relations, preprocessed text metadata for semantic searches, and entity identifiers for quicker entity lookups. Both graph and catalog databases are kept synchronized, and data modification is carried out exclusively in transaction mode.
The tool that implements the presented BioGraph model was created using Javascript programming language and NodeJS v19 runtime environment [
27]. The tool enables transforming and importing metadata from different data sources to objects and relations described in the model, indexing the catalog of imported entries, and executing queries for searching the stored data. The set of scripts used for metadata importing and transformation is extensible, and developers can easily include their own scripts for processing metadata from various sources. All import scripts are connected to the core BioGraph service, which exposes five essential methods for extracting model objects and relations and importing metadata. The developers use the exposed methods to start imports, map the metadata segments to objects and relations, and finish the import. The system can be deployed locally as a standalone system so that the queries can be executed offline.
The BioGraph system consists of importers, indexers, database connectors, API, and a core library, which connects all the pieces into a working system. Importers transform import data into data model objects using the methods exposed from the BioGraph core library. The core library maps the abstract model objects into graph nodes and edges that are stored in the graph database with the help of a graph database adapter. The core library also sends information about the received imports and objects to indexers, which store the data in the catalog database. There are four types of indexers: entry indexer, identifier indexer, description indexer, and import indexer. The identifier indexer stores identifiers in catalog tables indexed for quick keyword lookups, while description indexers store text descriptions of the objects in a similarly indexed table. The entry indexer logs the information about all stored objects and relations, while the import indexer logs all imports and links to objects that were imported during specific imports. The API allows external programs to access the BioGraph data with the help of a query parser, which transforms BioGraph queries into native language queries of the underlying graph database. The API specification is listed in the software documentation while the entire architecture of the BioGraph system is displayed in
Figure 4.
3.4. Data Flows
To give a complete picture of the BioGraph system,
Figure 4 shows enumerated steps for data transformation and retrieval, from importing data into the graph and catalog databases to fetching the query results in the Web user interface. The central group of services, shown in
Figure 4, enclosed in a blue dashed rectangle, is the core of the BioGraph system and it is static. Importers, enclosed in a green dashed rectangle, are dynamic services, that can be modified to fit any external data source. The first step of the process (step 1) is the collection of raw data from external databases. The data are downloaded using APIs of the external data sources, FTP access, or even scraping the web pages. The method for collecting the data is implemented into the import services, which can be customized for any data source. The subset of collected data is labeled within the import service and mapped to BioGraph objects and relations.
In the next step (step 2), labeled objects and relations go to the BioGraph Core service where the objects are first logged in the catalog database using indexers (step 3). Identifiers, descriptions, and general import information are indexed individually and stored in the catalog database (step 4). Duplicate entries are skipped, while non-duplicate objects and relations are sent to the graph database adapter (step 5). The graph database adapter transforms objects and relations into native storage queries of the underlying graph database and executes the queries on the graph database (step 6). New graph database adapters can be implemented for many of the existing graph database management systems. Storing both graph and catalog data is performed in transaction mode, to prevent data inconsistencies.
Once the data are stored in both graph and catalog databases, external applications, such as BioGraph Web UI can send BioGraph queries and keyword queries to the BioGraph API service in JSON form (step 7). The keyword queries are sent to indexers (step 8a) and executed on the catalog database (9b), while the parsed graph queries are transformed into native graph database queries in the graph database adapter (step 8b) and sent to the graph database (step 9b). Text data stored in the catalog database are internally stored in prefix trees to support the efficient execution of keyword queries.
The data flow once per import through the importers, with the exception of importing data updates, while the stored data can be queried arbitrarily many times using the core services.
3.5. Searching the Data
To remain database-agnostic, we designed a simple and generic internal query language that can be easily transformed into any of the native languages of the underlying database systems to support easy and efficient searches through metadata following the structure of the proposed model. The search is performed using query objects in JSON [
28] format, divided into two segments: “match” and “params”. The “match” segment lists the relations between the searched entities, while the “params” segment lists the identifiers and attributes of the searched objects and relations. An example of a query using internal BioGraph query language is shown in
Figure 5.
Users with technical experience can write their own applications to submit queries to BioGraph and use BioGraph as a backend system. Even though the integrated query language is simple and intuitive for users with technical backgrounds, it still may not be friendly enough for users with little or no technical experience. That is why we have also implemented a Web-based graphical user interface, developed using React framework, that communicates with BioGraph and can be used to create and execute graphical searches. It enables users to select predefined entity types and relations between the selected entities to draw a pattern that will be matched against the metadata graph. Graphical queries allow users without significant technical experience to intuitively create complex queries and discover indirect links between entities. Users can further explore the metadata of the entities from the query results and navigate through the graph following relations to neighboring entities. Additionally, the Web interface offers example queries and a help section to enable quicker onboarding of new users and a better understanding of the data retrieval process using graphical queries. All executed queries and results can be exported to files for further reuse and analysis. The file format for the exported queries is JSON, while the results can be exported in CSV format.
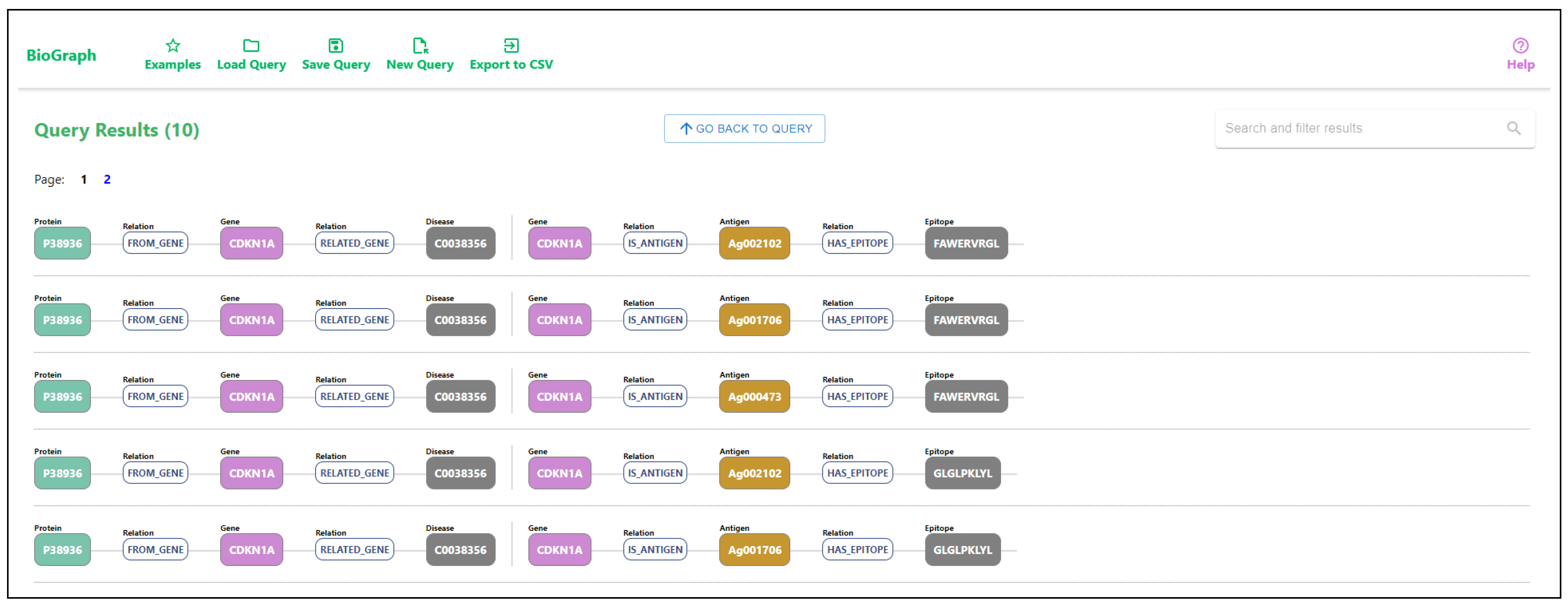
An example of a graphical query and the query results can be seen in
Figure 6. The graphical query describes a pattern connecting genes and related diseases with a relation score greater than 0.5, where the genes are also related to proteins with a disorder content percentage greater than 0.9. The relation score between the gene and the disease is the DisGeNET gene-disease association score [
20]. Additionally, we require that genes are also tumor antigens, and we want to match all epitopes related to those tumor antigens. Figure ref-results displays all matched patterns for the given graphical query. As the patterns are often not linear, we decompose the result patterns into linear disjoint paths. The results of the query are shown in
Figure 7, where the first result matches the example shown in
Figure 3.
Writing this type of query can be challenging in native database languages, while drawing a graphical pattern that the query should match is easy and intuitive. The results show paths from the matched patterns. Selecting any node from any path displays details about the specific entity and lists all the connected data and identifiers from all datasets. By following the relations listed in entity details, the user can easily traverse the graph and track the relations between the entities. An example of details for a specific gene is displayed in
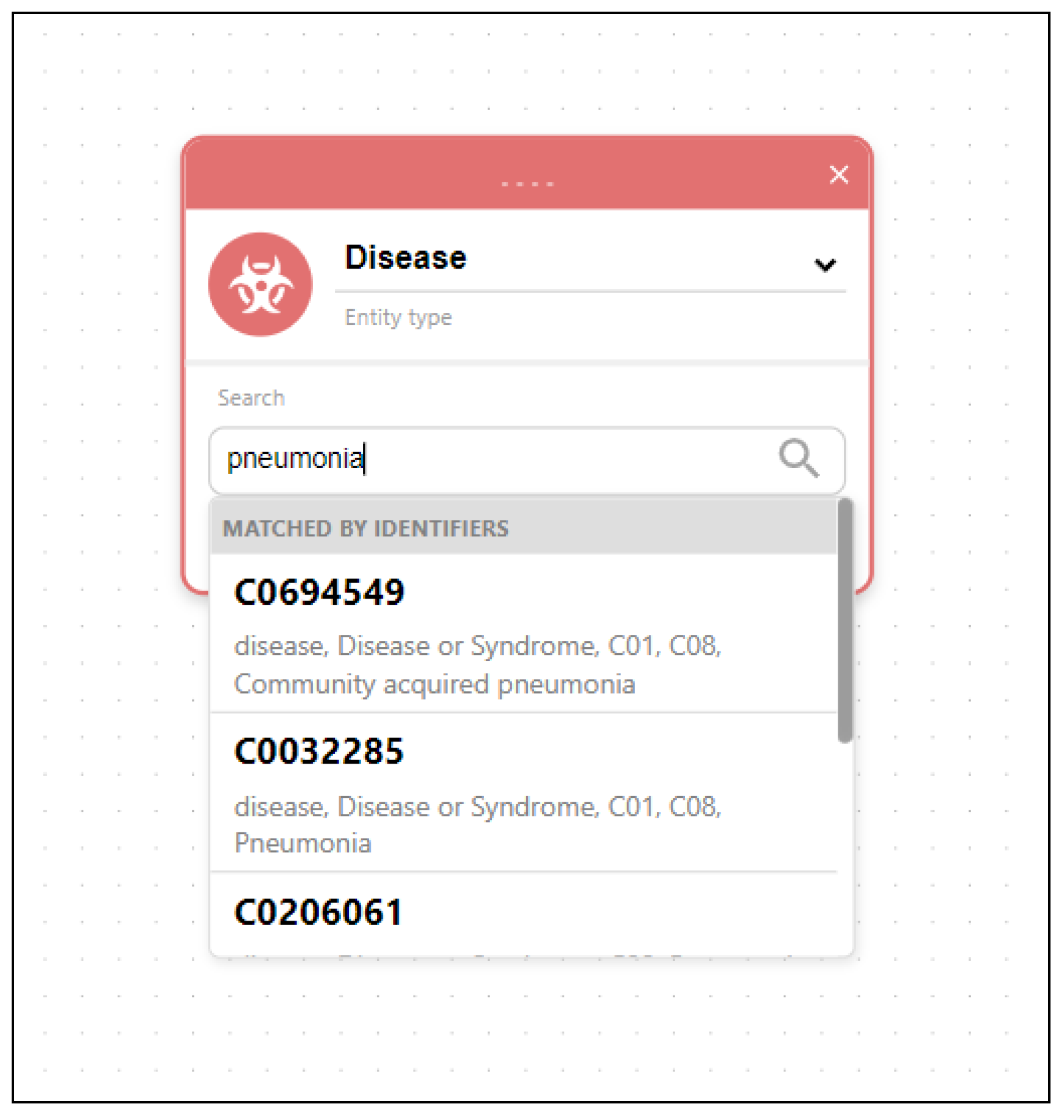
Figure 8. Besides using pattern matching, entities can also be searched using keyword searches through input fields located in entity nodes. An example of searching disease entity matching the keyword “pneumonia” is displayed in
Figure 9.
The BioGraph service and the Web interface are deployable in the local environment on Linux, Windows, and macOS platforms, requiring the installation of NodeJS runtime and Neo4J DBMS. The source code for the web interface, along with the source code of the Biograph services, can be found at
https://github.com/aleksandar-veljkovic/biograph (accessed on 25 February 2023). The predeployed BioGraph Web interface with all backend services preinstalled is currently available on
http://andromeda.matf.bg.ac.rs:54321 (accessed on 25 February 2023), but we highly encourage deploying the system in a local environment to maintain offline data availability.
3.6. Materials
For verification of the proposed model and its implementation, we collected metadata from five diversely formatted biological datasets: DisProt [
19], HGNC [
23], IEDB [
22], Tantigen 2.0 [
21], and DisGeNET [
20].
The DisProt (Database of Protein Disorder) dataset contains data on intrinsically disordered proteins from different species. The dataset currently consists of over 2300 protein entries. Metadata from the DisProt dataset were collected from DisProt API in JSON format.
The HGNC (HUGO Gene Nomenclature Committee, where HUGO stands for Human Genome Organization) dataset contains information on gene symbols, identifiers, gene chromosome locations, and their respective protein references for all known human genes. At the time of writing, the dataset contains 43,621 entries of human genes and also includes the identifiers of the ortholog genes found in mouse and rat genomes. Metadata from HGNC dataset were collected by downloading a JSON file.
The DisGeNet (Disease Gene Network) dataset contains information on the relations between genes and human diseases collected from multiple sources. The dataset consists of 1,134,942 gene-disease relation entries. The metadata from DisGeNET dataset were collected as a TSV document.
The IEDB (Immune Epitope Database) dataset contains data on immune epitopes from different organisms. Each epitope is associated with its source antigen, including identifier references to both protein and non-protein antigens, and source organism taxon. Although the dataset contains more information, such as those related to diseases, we decided for our model verification to use only the subset of information containing the relations between epitopes and antigens. The dataset metadata were downloaded from the project website in the TSV format.
The Tantigen 2.0 dataset contains data on human tumor antigens containing HLA ligands and immune T-cell epitopes. The database website does not provide API access or downloadable files. Our importer scraped the content of the project website and transformed the collected HTML pages into JSON documents as a preprocessing step. The total number of antigen entries in the Tantigen 2.0 dataset at the time of writing is 4297.
We have successfully transformed and imported and connected metadata from all datasets. The extracted metadata contained more than 16 million model objects, of which more than 2,500,000 individual entity objects, interconnected with more than 21 million relations. An example of the mapping of metadata from the DisGeNET dataset is provided in
Figure 10.
The system may be applied to new transcript descriptions in the databases for model plant species [
29,
30]. Querying the biological databases via web-interface was recently presented in the ANDDigest module integrated into the ANDSystem tool, designed to search for information in pre-processed texts [
31,
32]. The ontology of the ANDSystem tool features dictionaries for molecular-genetics entities (genes, proteins, metabolites, microRNAs), cells and organisms [
32], similar to our tool.
We have deployed the BioGraph services and the Web Interface Application to an Intel Xeon 3.50 GHz server with 16GB RAM running Windows 10 Server 2022 and Neo4J Community edition service. The data from the five datasets were preprocessed and imported into the graph and the catalog database in 10 h on the given machine. On a desktop workstation with 2.3 GHz Dual-Core Intel Core i5 and 8 GB RAM under MacOS Monterey, preprocessing and import time took 11 and a half hours. Still, the graph queries and searches by identifiers and keywords are executed in almost real-time (1–10 s), depending on query complexity.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}