
The oDGal Mouse: A Novel, Physiologically Relevant Rodent Model of Sporadic Alzheimer’s Disease
 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
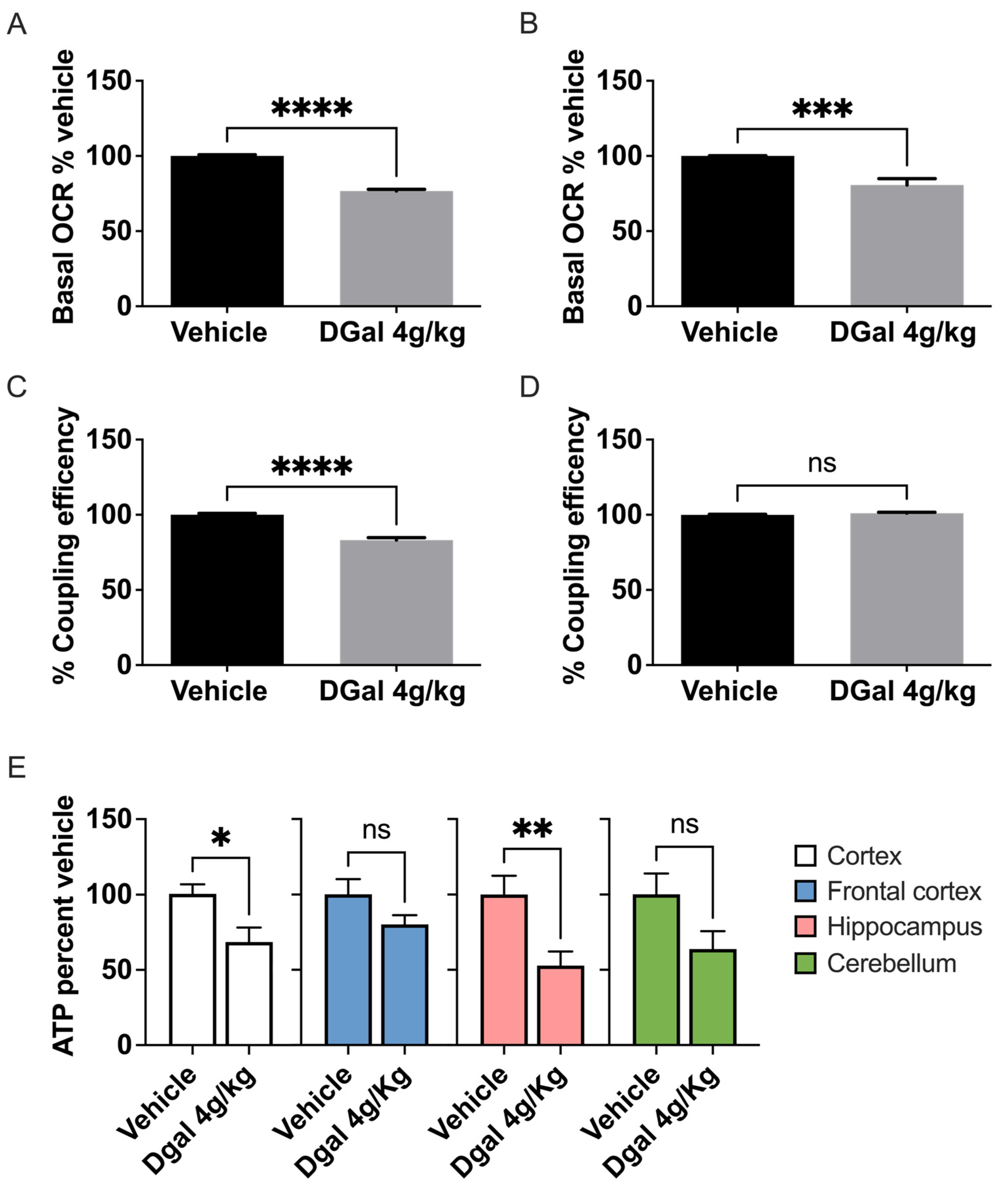
2.1. oDGal Induces Neuronal Hypometabolism
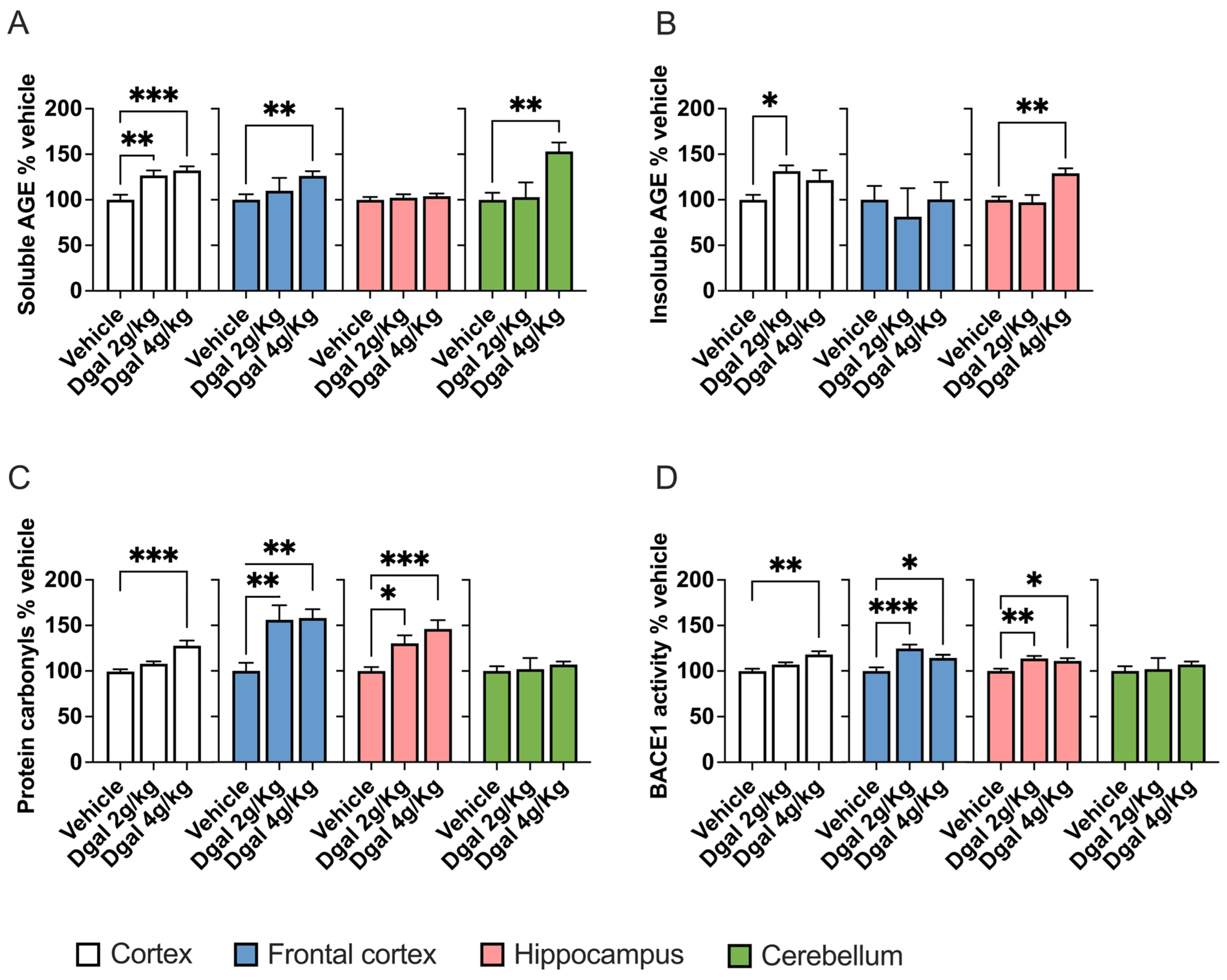
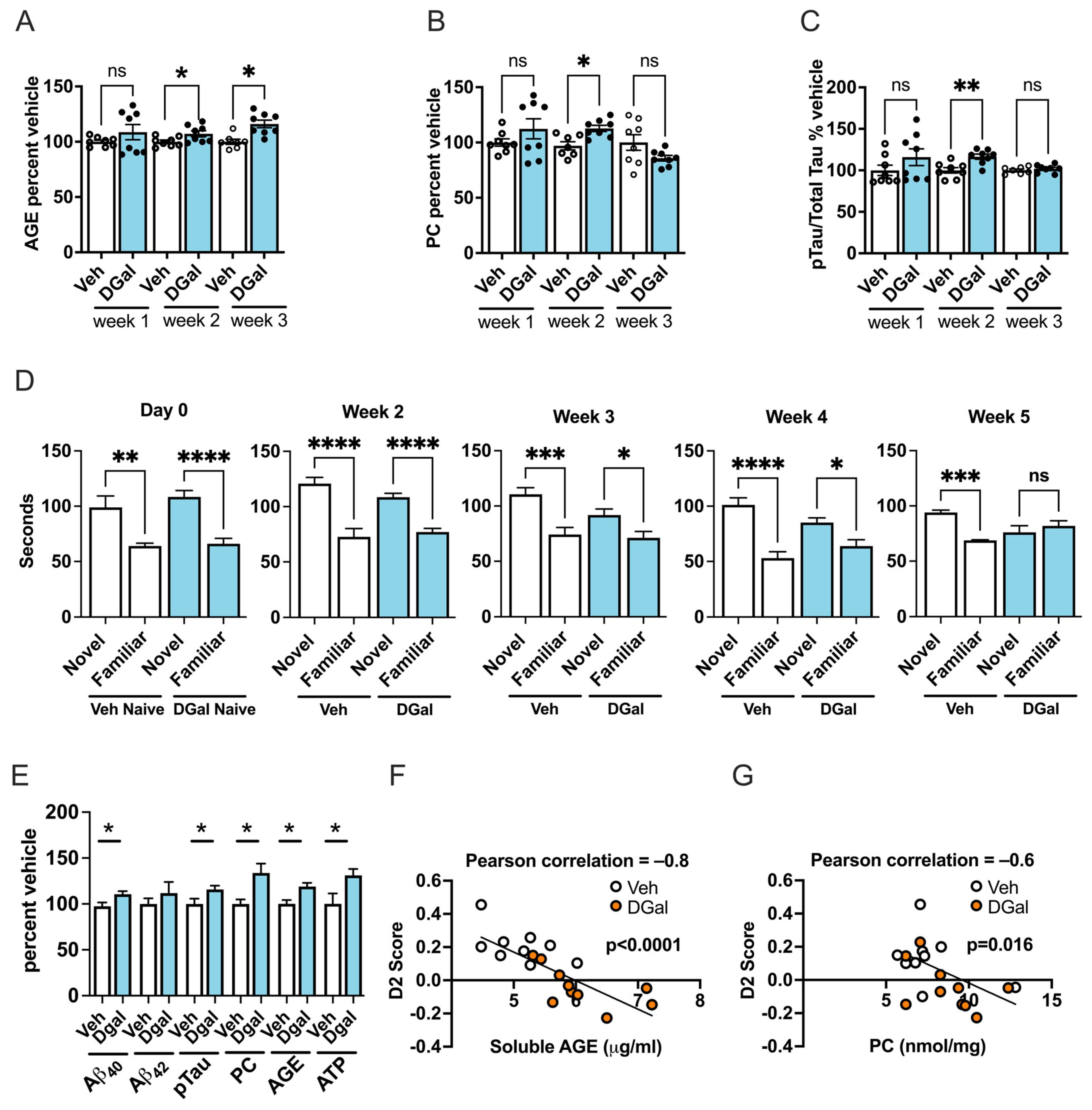
2.2. oDGal Increases AGE Products and Oxidative Stress
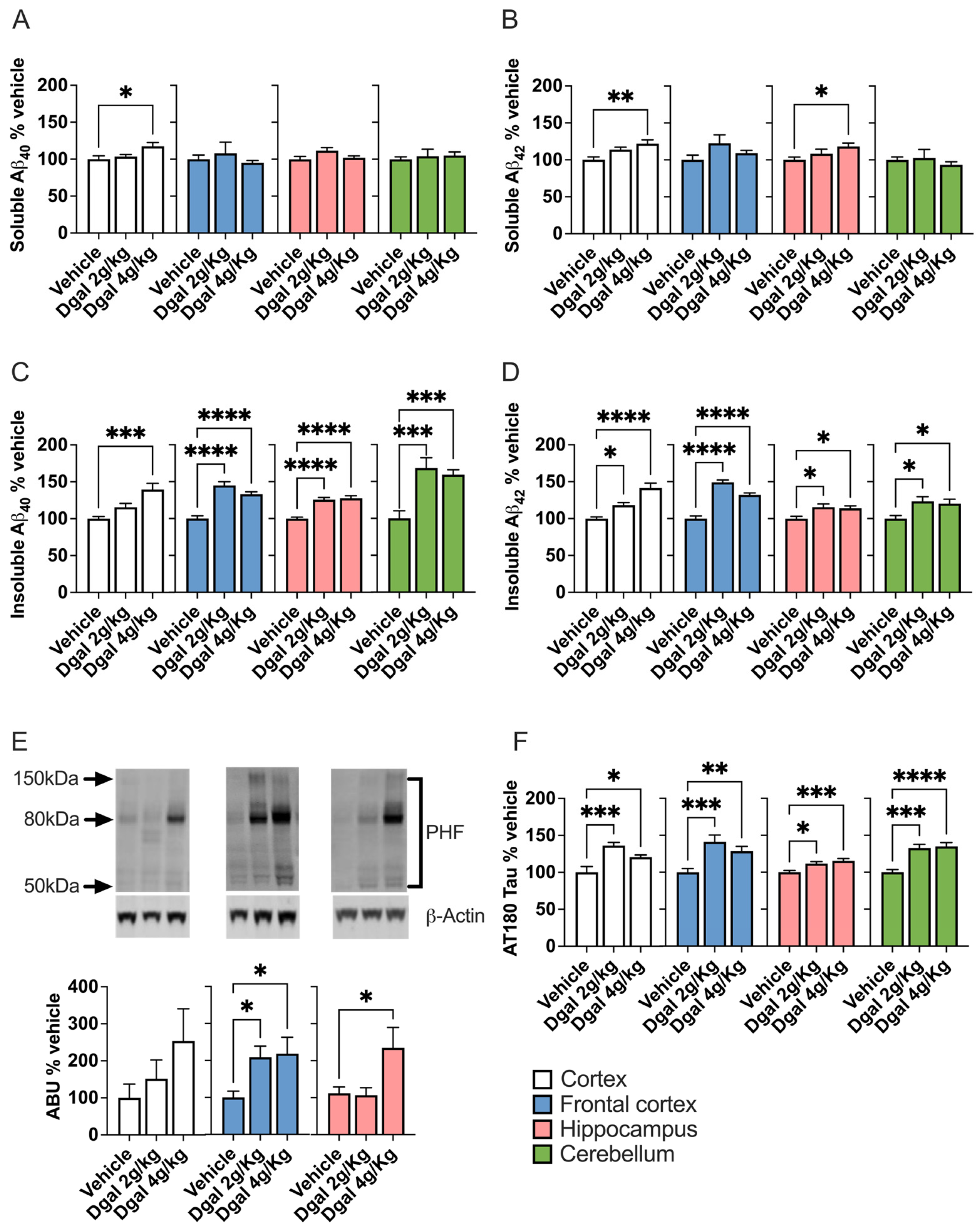
2.3. oDGal Increases Aβ and Hyperphosphorylated Tau
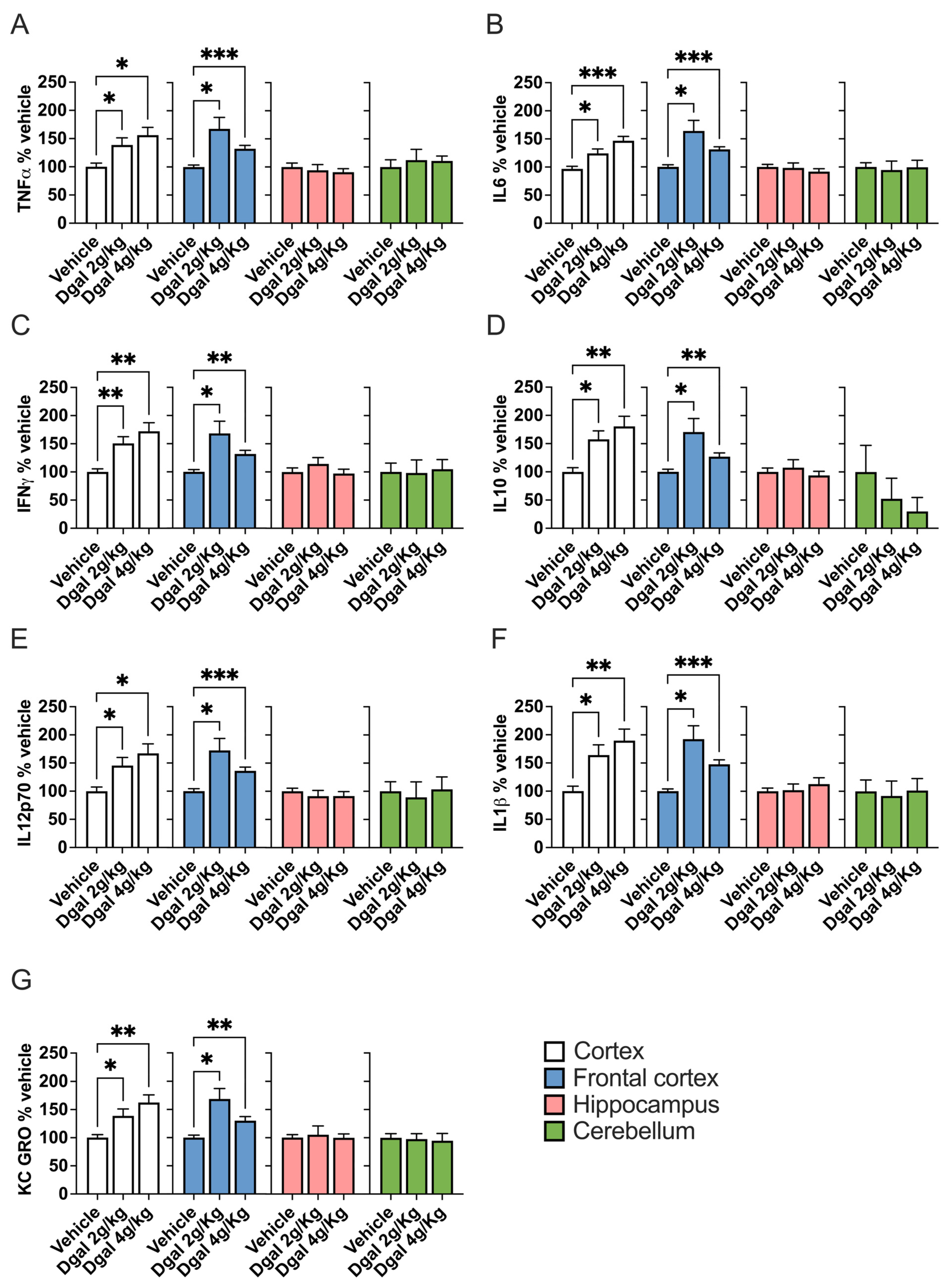
2.4. oDGal Promotes Neuroinflammation
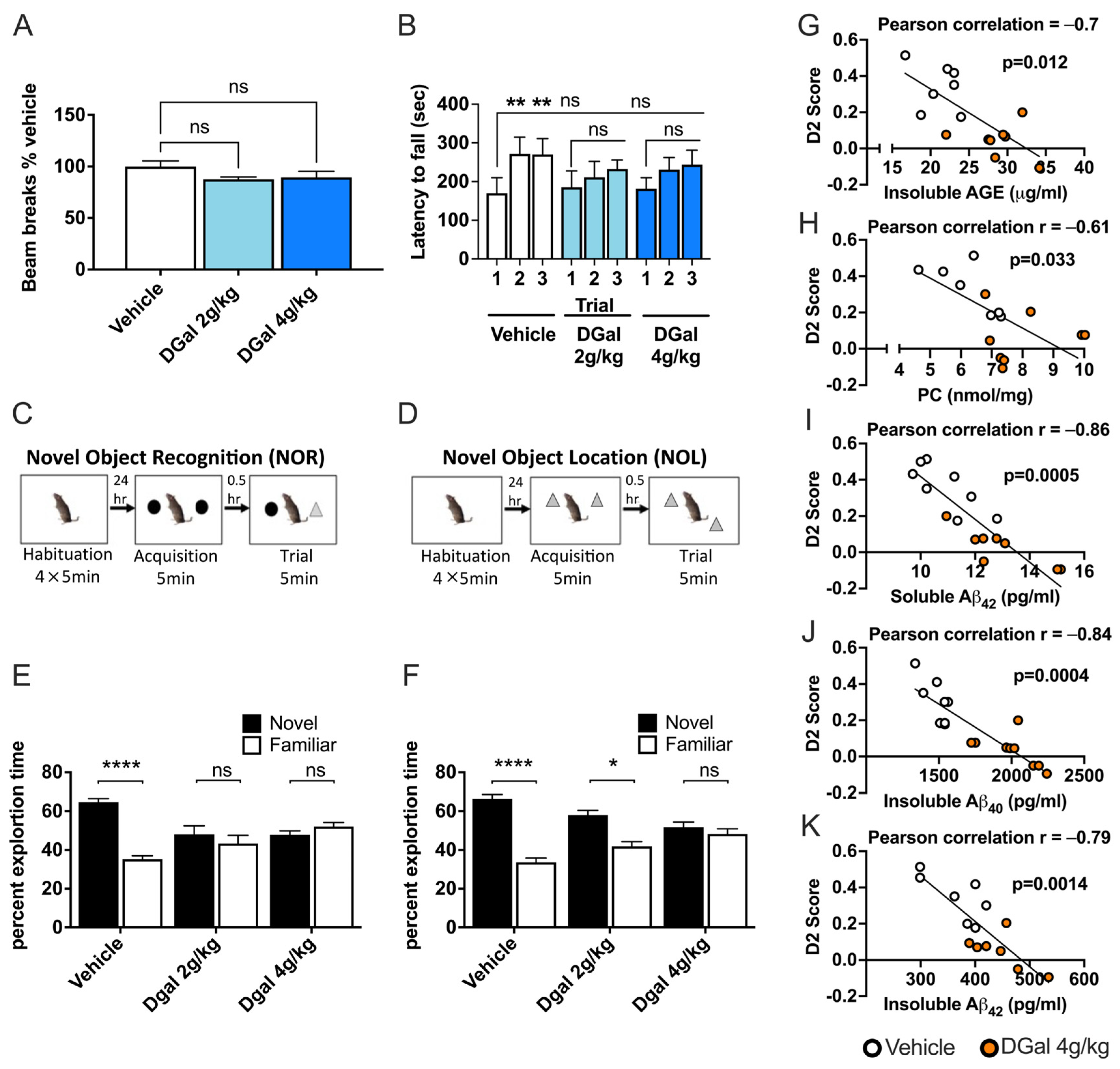
2.5. oDGal Affects Multiple Cognitive Domains
2.6. oDGal-Induced AGE, ROS, and Aβ Modulation Correlate with Spatial Memory Deficits
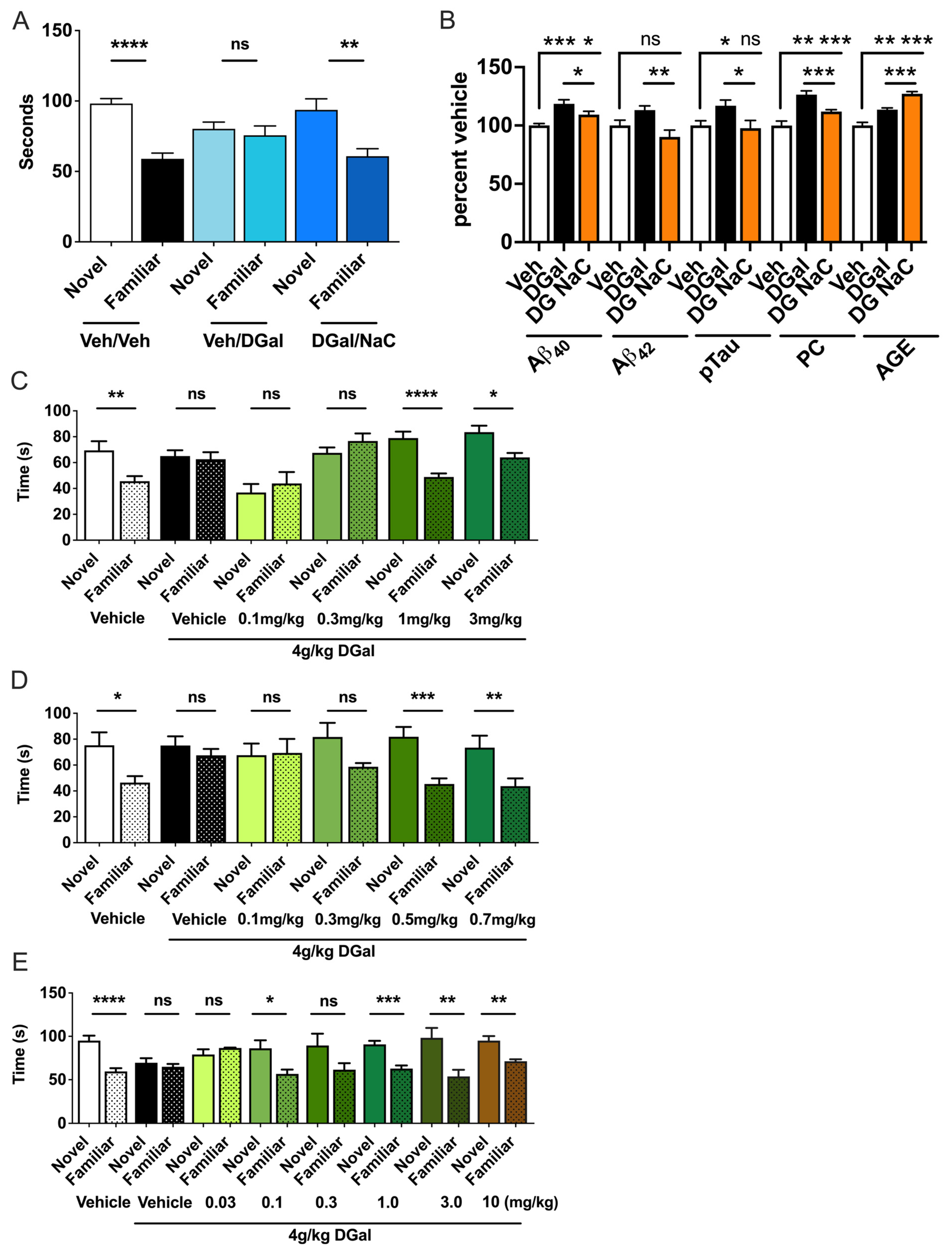
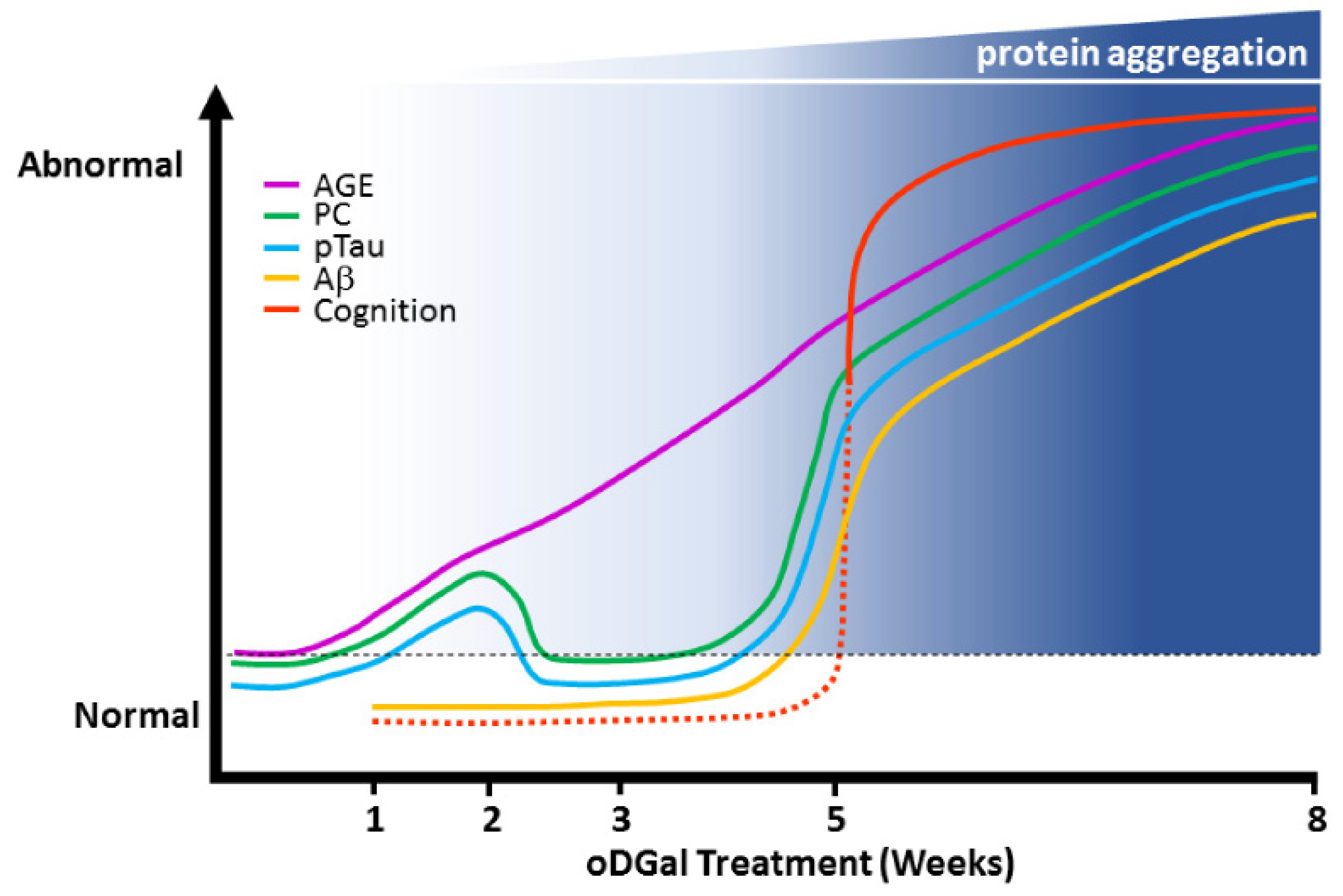
2.7. Etiology of oDGal Pathology
2.8. Therapeutic Interventions Ameliorate oDGal-Induced Pathology and Cognitive Decline
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. D-Galactose (oDGal) Treatment
4.3. oDGal Formulation
4.4. Synaptosome Preparation
4.5. Mitochondrial Functional Assay
4.6. Tissue Processing
4.7. ATP Assay
4.8. Protein Carbonyl and AGE Quantification
4.9. BACE1 Activity Assessment
4.10. Mesoscale Discovery (MSD) ELISA
4.11. Western Blot Analysis
4.12. Locomotor Activity Assay (LMA)
4.13. Rotarod Analyses
4.14. Novel Object Recognition (NOR)
4.15. Novel Object Location (NOL)
4.16. Spatial Recognition/Topographical Test
4.17. Drug Treatments
4.18. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Killin, L.O.J.; Starr, J.M.; Shiue, I.J.; Russ, T.C. Environmental risk factors for dementia: A systematic review. BMC Geriatr. 2016, 16, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic Risk Factors of Sporadic Alzheimer’s Disease: Implications in the Pathology, Pathogenesis and Treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Cong, W.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef]
- Maudsley, S.; Chadwick, W. Progressive and unconventional pharmacotherapeutic approaches to Alzheimer’s disease therapy. Curr. Alzheimer Res. 2012, 9, 1–4. [Google Scholar] [CrossRef]
- Chadwick, W.; Mitchell, N.; Martin, B.; Maudsley, S. Therapeutic targeting of the endoplasmic reticulum in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Geerts, H.; Dacks, P.A.; Devanarayan, V.; Haas, M.; Khachaturian, Z.S.; Gordon, M.F.; Maudsley, S.; Romero, K.; Stephenson, D. Brain Health Modeling Initiative (BHMI) Big data to smart data in Alzheimer’s disease: The brain health modeling initiative to foster actionable knowledge. Alzheimers Dement. 2016, 12, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Millan, M.J. The epigenetic dimension of Alzheimer’s disease: Causal, consequence, or curiosity? Dialogues Clin. Neurosci. 2014, 16, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef]
- Barnett, J.H.; Lewis, L.; Blackwell, A.D.; Taylor, M. Early intervention in Alzheimer’s disease: A health economic study of the effects of diagnostic timing. BMC Neurol. 2014, 14, 101. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Anastasiou, A.I.; Zachariou, V.; Pelidou, S.-H. Reasons for Failed Trials of Disease-Modifying Treatments for Alzheimer Disease and Their Contribution in Recent Research. Biomedicines 2019, 7, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, J.M.; Aisen, P.S.; Cummings, J.L.; Howard, R.J.; Fox, N.C. Unsuccessful trials of therapies for Alzheimer’s disease. Lancet 2019, 393, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; Lee, D.K.; Chung, B.-R.; Kim, H.V.; Kim, Y. Intracerebroventricular Injection of Amyloid-β Peptides in Normal Mice to Acutely Induce Alzheimer-like Cognitive Deficits. J. Vis. Exp. 2016, 109, e53308. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liang, Z.; Blanchard, J.; Dai, C.-L.; Sun, S.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; Liu, F.; Gong, C.-X. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: Similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 2013, 47, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimers Dis. 2010, 19, 311. [Google Scholar] [CrossRef] [Green Version]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Halliday, G. Pathology and hippocampal atrophy in Alzheimer’s disease. Lancet Neurol. 2017, 16, 862–864. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Li, X.-G.; Zhang, X.-Y.; Hou, J.-D.; Lin, L.-F.; Gao, Q.; Luo, H.-M. Combined administration of D-galactose and aluminium induces Alzheimer-like lesions in brain. Neurosci. Bull. 2011, 27, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Van Puyvelde, K.; Mets, T.; Njemini, R.; Beyer, I.; Bautmans, I. Effect of advanced glycation end product intake on inflammation and aging: A systematic review. Nutr. Rev. 2014, 72, 638–650. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmadi, A.; Steiner, N.; Münch, G. Advanced glycation endproducts as gerontotoxins and biomarkers for carbonyl-based degenerative processes in Alzheimer’s disease. Clin. Chem. Lab. Med. 2011, 49, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.; Oishi, N.; Ito, K.; Fukuyama, H.; SEAD-J Study Group. Alzheimer’s Disease Neuroimaging Initiative Effects of imaging modalities, brain atlases and feature selection on prediction of Alzheimer’s disease. J. Neurosci. Methods 2015, 256, 168–183. [Google Scholar] [CrossRef] [Green Version]
- Araque Caballero, M.Á.; Brendel, M.; Delker, A.; Ren, J.; Rominger, A.; Bartenstein, P.; Dichgans, M.; Weiner, M.W.; Ewers, M. Alzheimer’s Disease Neuroimaging Initative (ADNI) Mapping 3-year changes in gray matter and metabolism in Aβ-positive nondemented subjects. Neurobiol. Aging 2015, 36, 2913–2924. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Mancuso, C.; Di Domenico, F.; Sultana, R.; Murphy, M.P.; Head, E.; Butterfield, D.A. Biliverdin reductase-A: A novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 2012, 120, 135–146. [Google Scholar] [CrossRef]
- Sultana, R.; Reed, T.; Perluigi, M.; Coccia, R.; Pierce, W.M.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: A regional study. J. Cell. Mol. Med. 2007, 11, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Coccia, R.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimers Dis. 2011, 25, 623–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halverson, R.A.; Lewis, J.; Frausto, S.; Hutton, M.; Muma, N.A. Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J. Neurosci. 2005, 25, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflammation 2018, 15, 276. [Google Scholar] [CrossRef] [Green Version]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Barker, G.R.I.; Bird, F.; Alexander, V.; Warburton, E.C. Recognition memory for objects, place, and temporal order: A disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J. Neurosci. 2007, 27, 2948–2957. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-J.; Friston, K. Structural and Functional Brain Networks: From Connections to Cognition. Science 2013, 342, 1238411. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.E.; Sporns, O. Brain Networks and Cognitive Architectures. Neuron 2015, 88, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Medaglia, J.D.; Pasqualetti, F.; Hamilton, R.H.; Thompson-Schill, S.L.; Bassett, D.S. Brain and cognitive reserve: Translation via network control theory. Neurosci. Biobehav. Rev. 2017, 75, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Pasqualetti, G.; Tognini, S.; Calsolaro, V.; Polini, A.; Monzani, F. Potential drug–drug interactions in Alzheimer patients with behavioral symptoms. Clin. Interv. Aging 2015, 10, 1457–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagakura, A.; Shitaka, Y.; Yarimizu, J.; Matsuoka, N. Characterization of cognitive deficits in a transgenic mouse model of Alzheimer’s disease and effects of donepezil and memantine. Eur. J. Pharmacol. 2013, 703, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-Q.; Wang, B.-R.; Tian, Y.-Y.; Xu, J.; Gao, L.; Zhao, S.-L.; Jiang, T.; Xie, H.-G.; Zhang, Y.-D. Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci. Ther. 2013, 19, 871–881. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [Green Version]
- Kobro-Flatmoen, A.; Lagartos-Donate, M.J.; Aman, Y.; Edison, P.; Witter, M.P.; Fang, E.F. Re-emphasizing early Alzheimer’s disease pathology starting in select entorhinal neurons, with a special focus on mitophagy. Ageing Res. Rev. 2021, 67, 101307. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in Very Mild Alzheimer’s Disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef] [Green Version]
- Dickerson, B.C.; Feczko, E.; Augustinack, J.C.; Pacheco, J.; Morris, J.C.; Fischl, B.; Buckner, R.L. Differential effects of aging and Alzheimer’s disease on medial temporal lobe cortical thickness and surface area. Neurobiol. Aging 2009, 30, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Kulason, S.; Xu, E.; Tward, D.J.; Bakker, A.; Albert, M.; Younes, L.; Miller, M.I. Entorhinal and Transentorhinal Atrophy in Preclinical Alzheimer’s Disease. Front. Neurosci. 2020, 14, 804. [Google Scholar] [CrossRef]
- Weiler, M.; Agosta, F.; Canu, E.; Copetti, M.; Magnani, G.; Marcone, A.; Pagani, E.; Balthazar, M.L.F.; Comi, G.; Falini, A.; et al. Following the Spreading of Brain Structural Changes in Alzheimer’s Disease: A Longitudinal, Multimodal MRI Study. J. Alzheimers Dis. 2015, 47, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Raffaitin, C.; Gin, H.; Empana, J.-P.; Helmer, C.; Berr, C.; Tzourio, C.; Portet, F.; Dartigues, J.-F.; Alperovitch, A.; Barberger-Gateau, P. Metabolic Syndrome and Risk for Incident Alzheimer’s Disease or Vascular Dementia: The Three-City Study. Diabetes Care 2009, 32, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-T.; Chang, W.-N.; Tsai, N.-W.; Huang, C.-C.; Kung, C.-T.; Su, Y.-J.; Lin, W.-C.; Cheng, B.-C.; Su, C.-M.; Chiang, Y.-F.; et al. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer’s disease: A systematic review. BioMed Res. Int. 2014, 2014, 182303. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Friedland, R.P.; Hirai, K.; Chiba, S.; Smith, M.A. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J. Neuropathol. Exp. Neurol. 2000, 59, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Chiba, S.; Lippa, C.F.; Cras, P.; Kalaria, R.N.; Takeda, A.; Honda, K.; Smith, M.A.; Perry, G. Neuronal RNA oxidation is a prominent feature of familial Alzheimer’s disease. Neurobiol. Dis. 2004, 17, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Klunk, W.E. Early detection of Alzheimer’s disease using PiB and FDG PET. Neurobiol. Dis. 2014, 72 Pt A, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Minami, Y.; Sonoda, N.; Hayashida, E.; Makimura, H.; Ide, M.; Ikeda, N.; Ohgidani, M.; Kato, T.A.; Seki, Y.; Maeda, Y.; et al. p66Shc Signaling Mediates Diabetes-Related Cognitive Decline. Sci. Rep. 2018, 8, 3213. [Google Scholar] [CrossRef] [Green Version]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Yamagishi, S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, R.; Lu, S.; Xia, W.; Cai, R.; Sun, H.; Wang, S. RAGE and AGEs in Mild Cognitive Impairment of Diabetic Patients: A Cross-Sectional Study. PLoS ONE 2016, 11, e0145521. [Google Scholar] [CrossRef] [Green Version]
- Arnsten, A.F.T.; Datta, D.; Tredici, K.D.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Zetterberg, H.; Del Tredici, K.; Blennow, K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-β changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. 2013, 126, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Chen, G.; Cai, L.; Chen, B.; Liang, J.; Lin, F.; Li, L.; Lin, L.; Yao, J.; Wen, J.; Huang, H. Serum level of endogenous secretory receptor for advanced glycation end products and other factors in type 2 diabetic patients with mild cognitive impairment. Diabetes Care 2011, 34, 2586–2590. [Google Scholar] [CrossRef] [Green Version]
- Ghidoni, R.; Benussi, L.; Glionna, M.; Franzoni, M.; Geroldi, D.; Emanuele, E.; Binetti, G. Decreased plasma levels of soluble receptor for advanced glycation end products in mild cognitive impairment. J. Neural Transm. 2008, 115, 1047–1050. [Google Scholar] [CrossRef]
- Palmigiano, A.; Barone, R.; Sturiale, L.; Sanfilippo, C.; Bua, R.O.; Romeo, D.A.; Messina, A.; Capuana, M.L.; Maci, T.; Le Pira, F.; et al. CSF N-glycoproteomics for early diagnosis in Alzheimer’s disease. J. Proteomics 2016, 131, 29–37. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.Y.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Arslan, J.; Jamshed, H.; Qureshi, H. Early Detection and Prevention of Alzheimer’s Disease: Role of Oxidative Markers and Natural Antioxidants. Front. Aging Neurosci. 2020, 12, 231. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Gnjec, A.; Poon, H.F.; Castegna, A.; Pierce, W.M.; Klein, J.B.; Martins, R.N. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: An initial assessment. J. Alzheimers Dis. 2006, 10, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; De Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenini, G.; Sultana, R.; Memo, M.; Butterfield, D.A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 987–994. [Google Scholar] [CrossRef]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Guidi, I.; Galimberti, D.; Lonati, S.; Novembrino, C.; Bamonti, F.; Tiriticco, M.; Fenoglio, C.; Venturelli, E.; Baron, P.; Bresolin, N.; et al. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2006, 27, 262–269. [Google Scholar] [CrossRef]
- Xu, L.; Yang, Y.; Chen, J. The role of reactive oxygen species in cognitive impairment associated with sleep apnea. Exp. Ther. Med. 2020, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Kandlur, A.; Satyamoorthy, K.; Gangadharan, G. Oxidative Stress in Cognitive and Epigenetic Aging: A Retrospective Glance. Front. Mol. Neurosci. 2020, 13, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, J.P.; de Castro, A.A.; Soares, F.V.; da Cunha, E.F.F.; Ramalho, T.C. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, A.; Tamaoka, A.; Araki, W. Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J. Neurosci. Res. 2010, 88, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, I.; Ricny, J.; Atrakchi-Baranes, D.; Shemesh, C.; Kravitz, E.; Liraz-Zaltsman, S.; Maksin-Matveev, A.; Cooper, I.; Leibowitz, A.; Uribarri, J.; et al. High dietary advanced glycation end products are associated with poorer spatial learning and accelerated Aβ deposition in an Alzheimer mouse model. Aging Cell 2016, 15, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, G.; Mayer, S.; Michaelis, J.; Hipkiss, A.R.; Riederer, P.; Müller, R.; Neumann, A.; Schinzel, R.; Cunningham, A.M. Influence of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of beta-amyloid peptide. Biochim. Biophys. Acta 1997, 1360, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.; Lüth, H.J.; Deuther-Conrad, W.; Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Arendt, T.; Münch, G. Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer’s disease. Brain Res. 2001, 920, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.P.; Obrenovich, M.E.; Atwood, C.S.; Perry, G.; Smith, M.A. Involvement of Maillard reactions in Alzheimer disease. Neurotox. Res. 2002, 4, 191–209. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Wang, F.; Wang, J.; Liu, C.; Zhou, Y.; Xu, Z.; Zhang, C.; Sun, B.; Guan, Y. Pathological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: The Receptor for Advanced Glycation End Products (RAGE). Front. Aging Neurosci. 2020, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.-S.; Wu, M.-N.; Yang, C.-C.; Shen, C.-T.; Yang, Y.-H. Effect of Advanced Glycation End Products on the Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Ningappa, M.; Adenuga, M.; Ngo, K.A.; Mohamed, N.; Narayanan, T.; Prasadan, K.; Ashokkumar, C.; Das, J.; Schmitt, L.; Hartman, H.; et al. Mechanisms of Impaired Lung Development and Ciliation in Mannosidase-1-Alpha-2 (Man1a2) Mutants. Front. Physiol. 2021, 12, 658518. [Google Scholar] [CrossRef]
- Wang, R.; Xue, X.; Wang, Y.; Zhao, H.; Zhang, Y.; Wang, H.; Miao, D. BMI1 Deficiency Results in Female Infertility by Activating p16/p19 Signaling and Increasing Oxidative Stress. Int. J. Biol. Sci. 2019, 15, 870–881. [Google Scholar] [CrossRef]
- Kumar, V.; Jagadish, N.; Suri, A. Role of A-Kinase anchor protein (AKAP4) in growth and survival of ovarian cancer cells. Oncotarget 2017, 8, 53124–53136. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Xue, X.; Wang, Q.; Chen, N.; Miao, D. Bmi1 plays an important role in dentin and mandible homeostasis by maintaining redox balance. Am. J. Transl. Res. 2016, 8, 4716–4725. [Google Scholar]
- Buitrago, M.M.; Schulz, J.B.; Dichgans, J.; Luft, A.R. Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol. Learn. Mem. 2004, 81, 211–216. [Google Scholar] [CrossRef]
- Lalonde, R.; Hayzoun, K.; Selimi, F.; Mariani, J.; Strazielle, C. Motor coordination in mice with hotfoot, Lurcher, and double mutations of the Grid2 gene encoding the delta-2 excitatory amino acid receptor. Physiol. Behav. 2003, 80, 333–339. [Google Scholar] [CrossRef]
- Rozas, G.; Guerra, M.J.; Labandeira-García, J.L. An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res. Protoc. 1997, 2, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, K.; Mulder, G.B. The rotarod. Contemp. Top. Lab. Anim. Sci. 2003, 42, 49. [Google Scholar] [PubMed]
- Rustay, N.R.; Wahlsten, D.; Crabbe, J.C. Influence of task parameters on rotarod performance and sensitivity to ethanol in mice. Behav. Brain Res. 2003, 141, 237–249. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chadwick, W.; Maudsley, S.; Hull, W.; Havolli, E.; Boshoff, E.; Hill, M.D.W.; Goetghebeur, P.J.D.; Harrison, D.C.; Nizami, S.; Bedford, D.C.; et al. The oDGal Mouse: A Novel, Physiologically Relevant Rodent Model of Sporadic Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 6953. https://doi.org/10.3390/ijms24086953
Chadwick W, Maudsley S, Hull W, Havolli E, Boshoff E, Hill MDW, Goetghebeur PJD, Harrison DC, Nizami S, Bedford DC, et al. The oDGal Mouse: A Novel, Physiologically Relevant Rodent Model of Sporadic Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(8):6953. https://doi.org/10.3390/ijms24086953
Chicago/Turabian StyleChadwick, Wayne, Stuart Maudsley, William Hull, Enes Havolli, Eugene Boshoff, Mark D. W. Hill, Pascal J. D. Goetghebeur, David C. Harrison, Sohaib Nizami, David C. Bedford, and et al. 2023. "The oDGal Mouse: A Novel, Physiologically Relevant Rodent Model of Sporadic Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 8: 6953. https://doi.org/10.3390/ijms24086953