
Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor Ameliorate Metabolic Disorder and Obesity Induced Cardiomyocyte Injury and Mitochondrial Remodeling
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. The Epicardial Fat Secretome Induces Cardiotoxicity and Mitochondrial Dysfunction
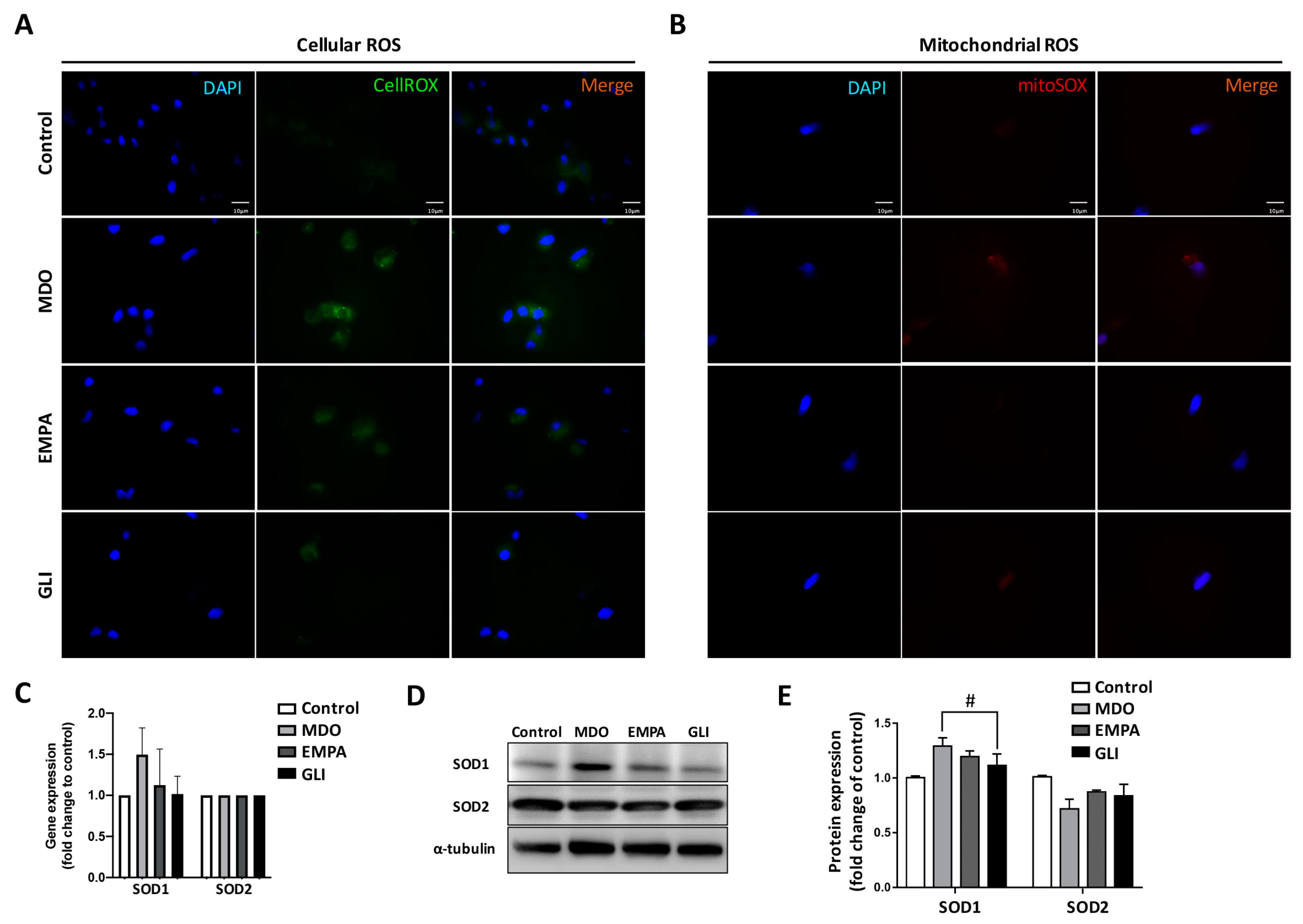
2.2. Mitochondrial Dysregulation Leads to Increased Intracellular ROS Production
2.3. Effects of the EMPA Secretome on Mitochondrial Function Regulators
2.4. RNA-Seq Analysis of the Effects of the EMPA Secretome on H9c2 Cells
2.5. Verification of the Effects of the EMPA Secretome on the Myocardial Ventricle in the MDO Mouse Model
3. Discussion
Limitations
4. Materials and Methods
4.1. Study Animal Preparation
4.2. Epicardial Fat Secretome Collection
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Real-Time PCR Analysis
4.6. Western Blot Analysis
4.7. Immunohistological Analysis
4.8. RNA Sequencing
4.9. Bioinformatic Analysis and IPA/KEGG/STRING Analyses
4.10. Mitochondrial Function Evaluation by JC-1 Assay
4.11. Mitochondrial Adenosine Triphosphate (ATP) Production
4.12. Mitochondrial NADH/NAD+ Synthesis
4.13. ROS Investigation Assay
4.14. Determination of Mitochondrial Ca2+ Distribution
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asghar, O.; Alam, U.; Hayat, S.A.; Aghamohammadzadeh, R.; Heagerty, A.M.; Malik, R.A. Obesity, diabetes and atrial fibrillation; epidemiology, mechanisms and interventions. Curr. Cardiol. Rev. 2012, 8, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. Obesity and cardiovascular disease: A scientific statement from the american heart association. Circulation 2021, 143, e984–e1010. [Google Scholar] [CrossRef]
- Tam, W.C.; Lin, Y.K.; Chan, W.P.; Huang, J.H.; Hsieh, M.H.; Chen, S.A.; Chen, Y.J. Pericardial fat is associated with the risk of ventricular arrhythmia in asian patients. Circ. J. Off. J. Jpn. Circ. Soc. 2016, 80, 1726–1733. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Zhang, S.; Yang, D.; Gao, L.; Lin, Y.; Chu, Z.; Jiang, X.; Yin, X.; Zheng, Z.; Wei, X.; et al. Effect of epicardial fat pad ablation on acute atrial electrical remodeling and inducibility of atrial fibrillation. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.T.; Tang, P.W.; Tsai, W.C.; Liu, I.H.; Yen, H.W.; Voon, W.C.; Wu, B.N.; Sheu, S.H.; Lai, W.T. Differential effects of central and peripheral fat tissues on the delayed rectifier k(+) outward currents in cardiac myocytes. Cardiology 2013, 125, 118–124. [Google Scholar] [CrossRef]
- Jhuo, S.J.; Liu, I.H.; Tsai, W.C.; Chou, T.W.; Lin, Y.H.; Wu, B.N.; Lee, K.T.; Lai, W.T. Effects of secretome from fat tissues on ion currents of cardiomyocyte modulated by sodium-glucose transporter 2 inhibitor. Molecules 2020, 25, 3606. [Google Scholar] [CrossRef]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing sglt2 as a therapeutic target for diabetes: Basic physiology and consequences. Diabetes Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Jhuo, S.J.; Lin, T.H.; Lin, Y.H.; Tsai, W.C.; Liu, I.H.; Wu, B.N.; Lee, K.T.; Lai, W.T. Clinical observation of sglt2 inhibitor therapy for cardiac arrhythmia and related cardiovascular disease in diabetic patients with controlled hypertension. J. Pers. Med. 2022, 12, 271. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. Sglt2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. Nadph oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Kushnir, A.; Zalk, R.; Yuan, Q.; Melville, Z.; Marks, A.R. Intracellular calcium leak in heart failure and atrial fibrillation: A unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 2020, 17, 732–747. [Google Scholar] [CrossRef]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic. Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via ampk-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovasc. Diabetol. 2020, 19, 62. [Google Scholar] [CrossRef]
- Xu, L.; Xu, C.; Liu, X.; Li, X.; Li, T.; Yu, X.; Xue, M.; Yang, J.; Kosmas, C.E.; Moris, D.; et al. Empagliflozin induces white adipocyte browning and modulates mitochondrial dynamics in kk cg-ay/j mice and mouse adipocytes. Front. Physiol. 2021, 12, 745058. [Google Scholar] [CrossRef]
- Morciano, G.; Vitto, V.A.M.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial bioenergetics and dynamism in the failing heart. Life 2021, 11, 436. [Google Scholar] [CrossRef]
- Ramalingam, A.; Budin, S.B.; Mohd Fauzi, N.; Ritchie, R.H.; Zainalabidin, S. Targeting mitochondrial reactive oxygen species-mediated oxidative stress attenuates nicotine-induced cardiac remodeling and dysfunction. Sci. Rep. 2021, 11, 13845. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. Nlrp3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudina, S.; Bugger, H.; Sena, S.; O’Neill, B.T.; Zaha, V.G.; Ilkun, O.; Wright, J.J.; Mazumder, P.K.; Palfreyman, E.; Tidwell, T.J.; et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009, 119, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Sodhi, K.; Puri, N.; Monu, S.R.; Rezzani, R.; Abraham, N.G. High fat diet enhances cardiac abnormalities in shr rats: Protective role of heme oxygenase-adiponectin axis. Diabetol. Metab. Syndr. 2011, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Xu, J.; Zou, X.; Li, Y.; Chen, C.; Zheng, A.; Li, H.; Li, H.; Szeto, I.M.; Shi, Y.; et al. Hydroxytyrosol prevents diet-induced metabolic syndrome and attenuates mitochondrial abnormalities in obese mice. Free. Radic. Biol. Med. 2014, 67, 396–407. [Google Scholar] [CrossRef] [PubMed]
- González, M.; Rojas, S.; Avila, P.; Cabrera, L.; Villalobos, R.; Palma, C.; Aguayo, C.; Peña, E.; Gallardo, V.; Guzmán-Gutiérrez, E.; et al. Insulin reverses d-glucose-increased nitric oxide and reactive oxygen species generation in human umbilical vein endothelial cells. PLoS ONE 2015, 10, e0122398. [Google Scholar] [CrossRef] [Green Version]
- Gill, A.; Gray, S.P.; Jandeleit-Dahm, K.A.; Watson, A.M.D. Sglt-2 inhibition: Novel therapeutics for reno-and cardioprotection in diabetes mellitus. Curr. Diabetes Rev. 2019, 15, 349–356. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Rodriguez-Sinovas, A.; Girao, H. Cellular crosstalk in cardioprotection: Where and when do reactive oxygen species play a role? Free. Radic. Biol. Med. 2021, 169, 397–409. [Google Scholar] [CrossRef]
- Jhuo, S.J.; Liu, I.H.; Tasi, W.C.; Chou, T.W.; Lin, Y.H.; Wu, B.N.; Lee, K.T.; Lai, W.T. Characteristics of ventricular electrophysiological substrates in metabolic mice treated with empagliflozin. Int. J. Mol. Sci. 2021, 22, 6105. [Google Scholar]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Xue, P.; Zhong, B.; Mao, A.P.; Ran, Y.; Chen, H.; Wang, Y.Y.; Yang, F.; Shu, H.B. Isg56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. USA 2009, 106, 7945–7950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawowczyk, M.; Van Scoy, S.; Kumar, K.P.; Reich, N.C. The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem. 2011, 286, 7257–7266. [Google Scholar] [CrossRef] [Green Version]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium signaling and cardiac arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Vassalle, M.; Lin, C.I. Calcium overload and cardiac function. J. Biomed. Sci. 2004, 11, 542–565. [Google Scholar] [CrossRef] [PubMed]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. Jc-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell. Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuck, B.J.; Lenski, M.; Böhm, M.; Laufs, U. Metabolic switch and hypertrophy of cardiomyocytes following treatment with angiotensin ii are prevented by amp-activated protein kinase. J. Biol. Chem. 2008, 283, 32562–32569. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward | Reserve |
|---|---|---|
| mfn1 | TCAGAGCCTATCTTTCAGCTCC | ACGGACGCCAATCCTGTTAC |
| mfn2 | AGCAAAGCTGCTCAGGAATAAA | CCTCTCCACGCAGACTCC |
| mx1 | CACACCGTGACGGATATGGT | TTTGGACTTGGCGGTTCTGT |
| mx2 | TGAACGTGCAGCGAGCTT | GGCTTGTGGGCCTTAGACAT |
| zbp1 | ACGATTTACCGCCCAGAAGA | TCCAGCTGTTGGGTCCATTC |
| ifit1 | ACAGCAACCATGAGGTTCTTTA | CCACAAGACATCAGAGAGGCT |
| ifit1b | CTTCATAGCACCATGAGTGAAGA | CAGGAATTTCAGGGGCTTCAA |
| oasl | CTGAAGGTAGTCAAGGTGGGC | TTTGTGATGCTTGGCTGCCT |
| isg15 | ACAGCCATGGGCTGGGA | CCTTCAGCTCTGACACCGAC |
| RYR1 | TTCTTCCCTGTCGTGAGCTT | CATGGAGCATAGCCAGGTG |
| RYR2 | TTTTCTGGTTCCCTCGACTG | TTCAGAGCTTCTGGGCTCTC |
| MCU | AATTAAAGCATTGCAGGTGGA | CCCTTCTTCCCTCAGATCCT |
| MICU1 | GACCCGGTAGATGGGAGAAT | CTTCAGCTGTCTCTGCATGG |
| MCUR1 | TGCAAACTGTGGATCCGTAA | TTGAAAGTGTGGTCCATCCA |
| VDAC1 | TGGCTCCATTTACCAGAAGG | CGACCTGATACTTGGCTGCT |
| VDAC2 | CTGGGGACTTCCAGCTACAC | CCTGATGTCCAAGCAAGGTT |
| VDAC3 | TTGACACAGCCAAATCCAAA | CTCCAAACTCAGTGCCATCA |
| Bax | TGTTTGCTGATGGCAACTTC | GATCAGCTCGGGCACTTTAG |
| Bcl2 | AAGCTGTCACAGAGGGGCTA | CTCTCAGGCTGGAAGGAGAA |
| ANT1 | GGCGACTGTCTCACCAAGAT | TCCGAAGTAGGCAGCTCTGT |
| ANT2 | CAGCTGGATGATTGCACAGT | ATCAGTTCCTTTGCGTCCAG |
| ANT3 | CAGCTGGATGATTGCACAGT | TCAGTTCTTTTGCGTCCAGA |
| CypD-PPID | AGGACTAGGTGTGGCAAGGA | CCAGTCATCCCCTTCTTTCA |
| CypD-PPIF | GTCAAAGAGGGCATGGATGT | AGCTCAACTGGCCACAGTCT |
| MnSOD(SOD2) | TGGTGCCTCTGGGTTTTCTA | ATCGGACAGGCCCTACCTAC |
| SOD1 | GAGACCTGGGCAATGTGACT | TCATGGACCACCATTGTACG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jhuo, S.-J.; Lin, Y.-H.; Liu, I.-H.; Lin, T.-H.; Wu, B.-N.; Lee, K.-T.; Lai, W.-T. Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor Ameliorate Metabolic Disorder and Obesity Induced Cardiomyocyte Injury and Mitochondrial Remodeling. Int. J. Mol. Sci. 2023, 24, 6842. https://doi.org/10.3390/ijms24076842
Jhuo S-J, Lin Y-H, Liu I-H, Lin T-H, Wu B-N, Lee K-T, Lai W-T. Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor Ameliorate Metabolic Disorder and Obesity Induced Cardiomyocyte Injury and Mitochondrial Remodeling. International Journal of Molecular Sciences. 2023; 24(7):6842. https://doi.org/10.3390/ijms24076842
Chicago/Turabian StyleJhuo, Shih-Jie, Yi-Hsiung Lin, I-Hsin Liu, Tsung-Hsien Lin, Bin-Nan Wu, Kun-Tai Lee, and Wen-Ter Lai. 2023. "Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor Ameliorate Metabolic Disorder and Obesity Induced Cardiomyocyte Injury and Mitochondrial Remodeling" International Journal of Molecular Sciences 24, no. 7: 6842. https://doi.org/10.3390/ijms24076842