
Transcriptomic Differences Underlying the Activin-A Induced Large Osteoclast Formation in Both Healthy Control and Fibrodysplasia Ossificans Progressiva Osteoclasts
 , , , and
, , , and
Abstract
:1. Introduction
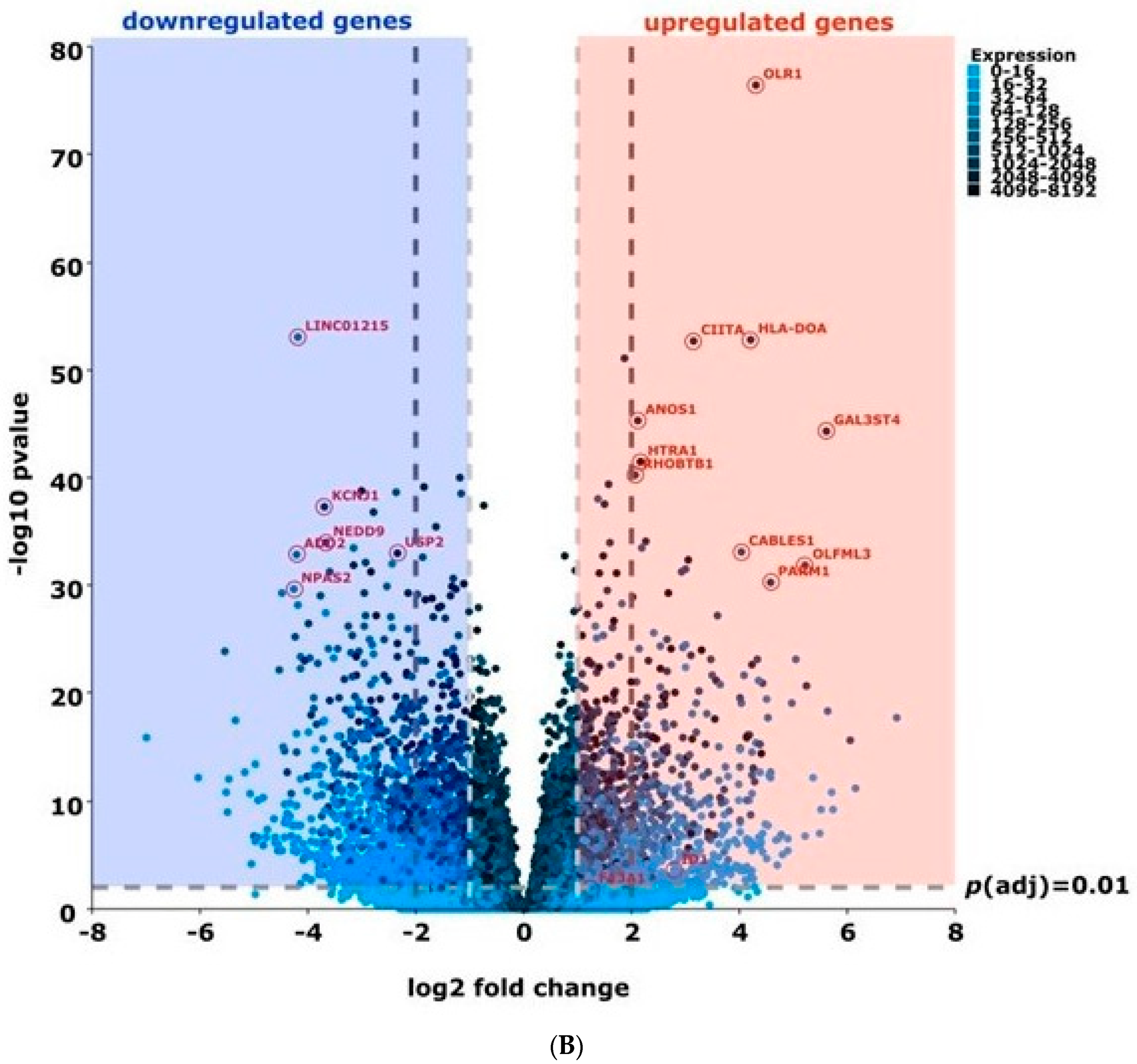
2. Results
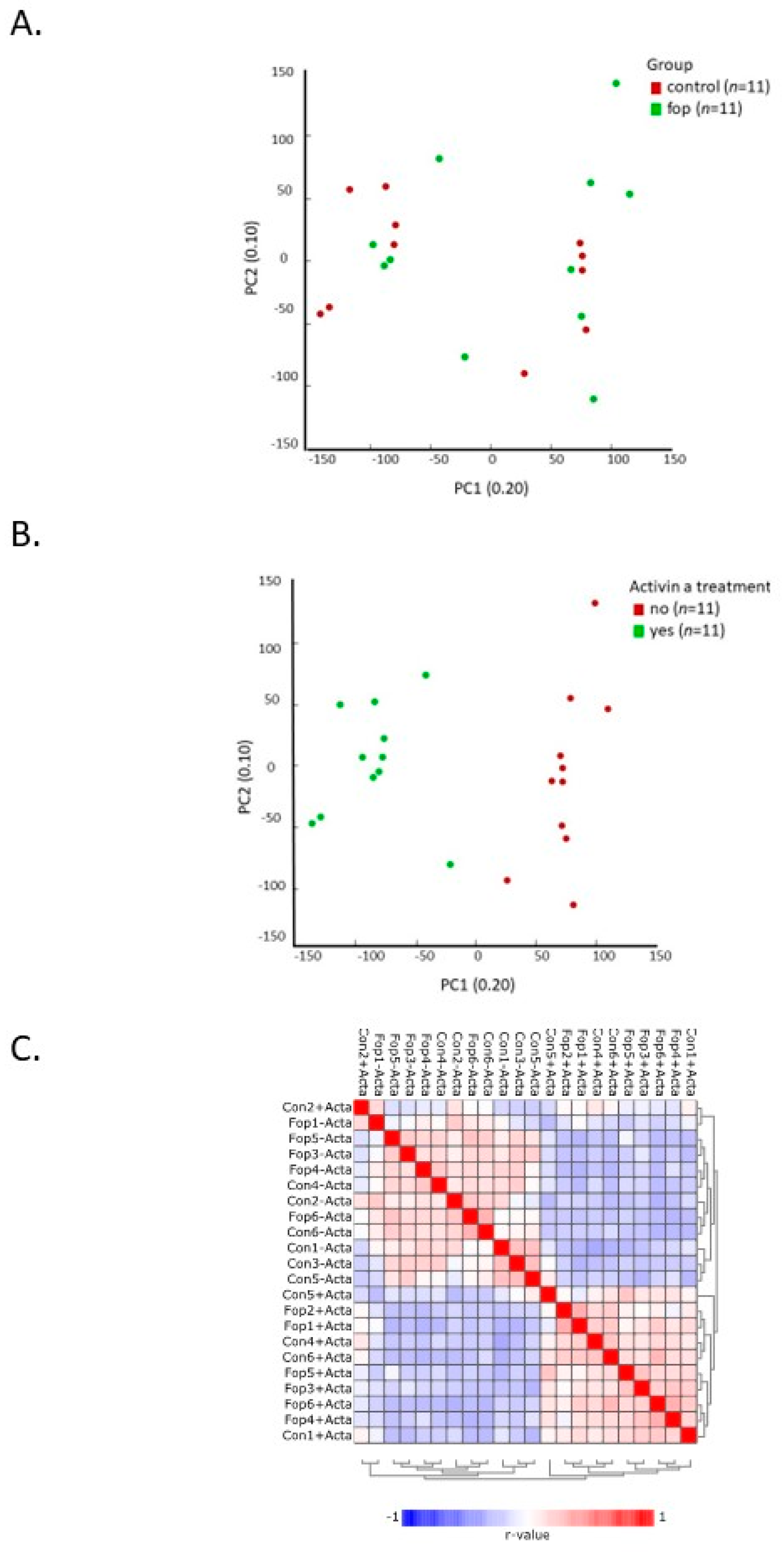
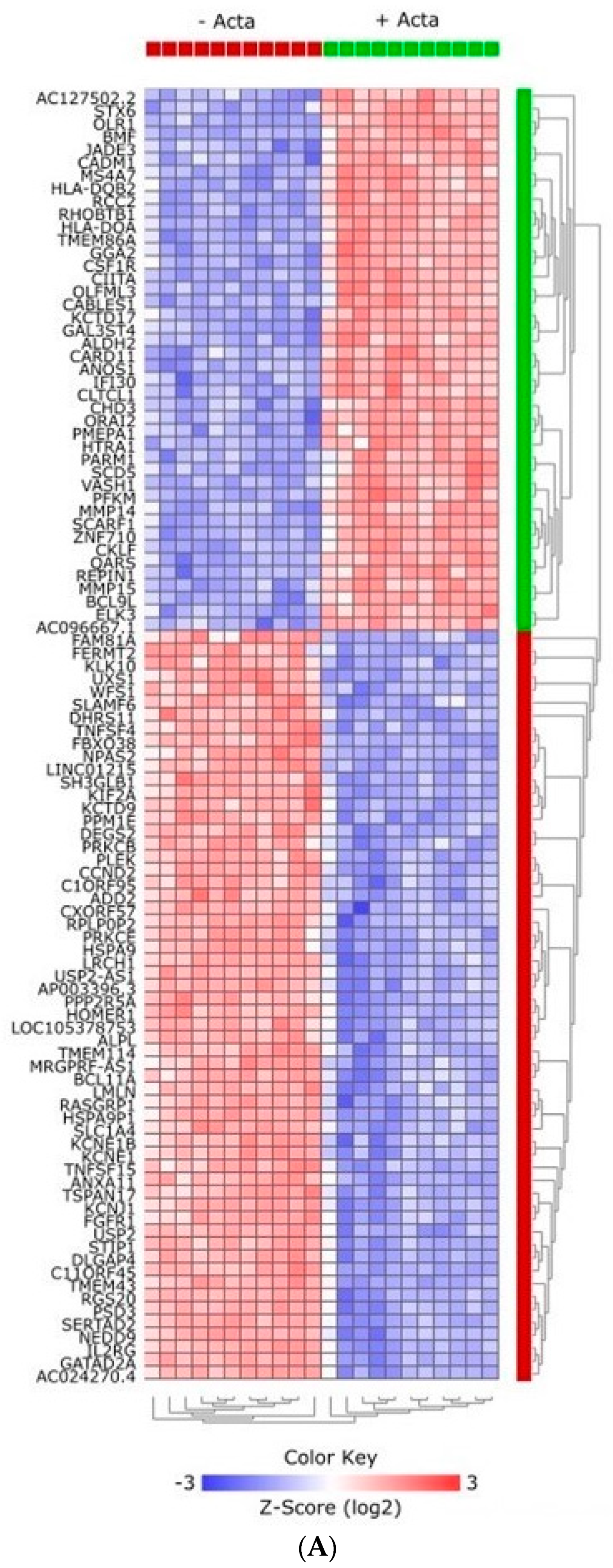
2.1. Experimental Treatment Samples Clustered Together
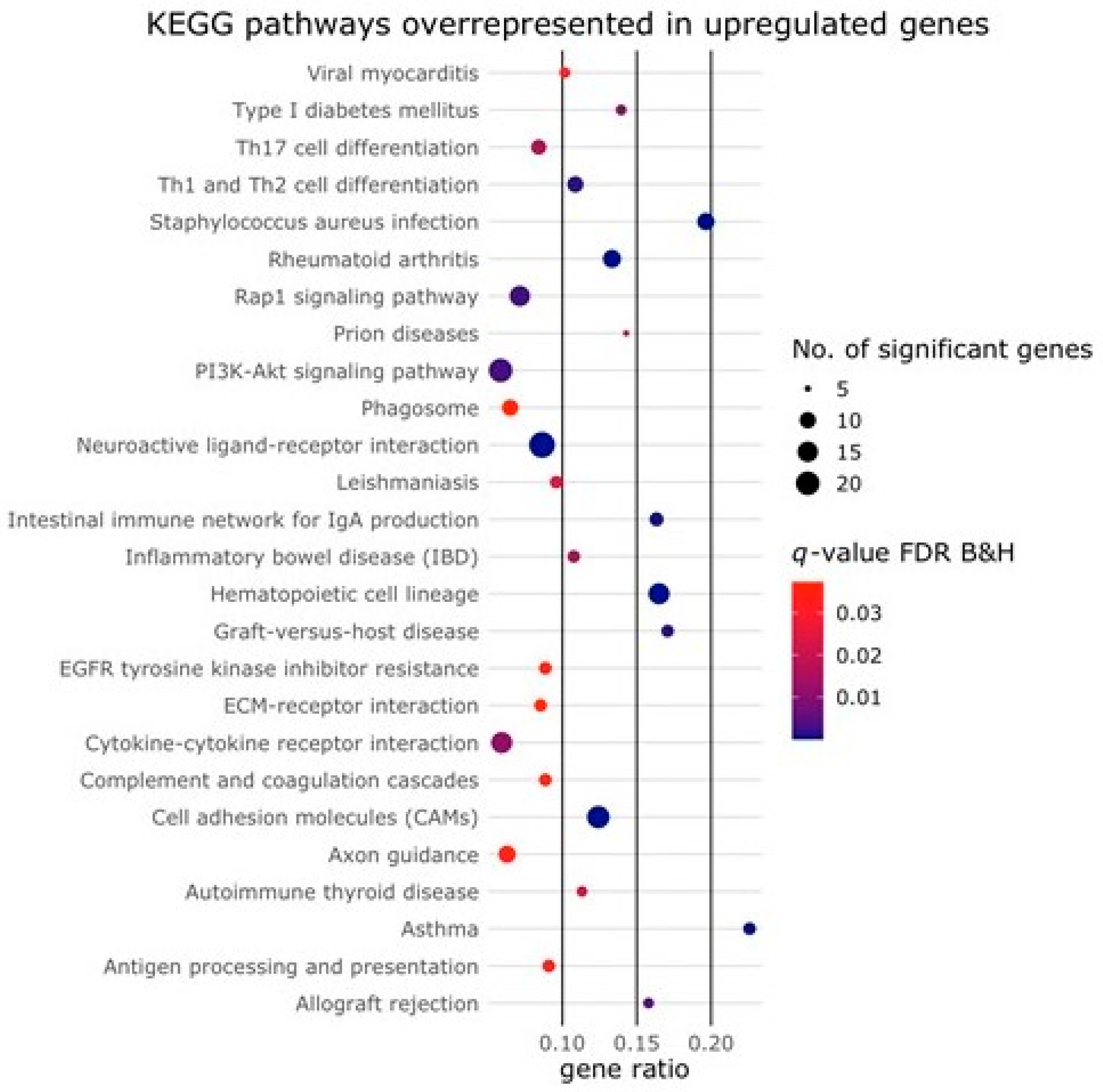
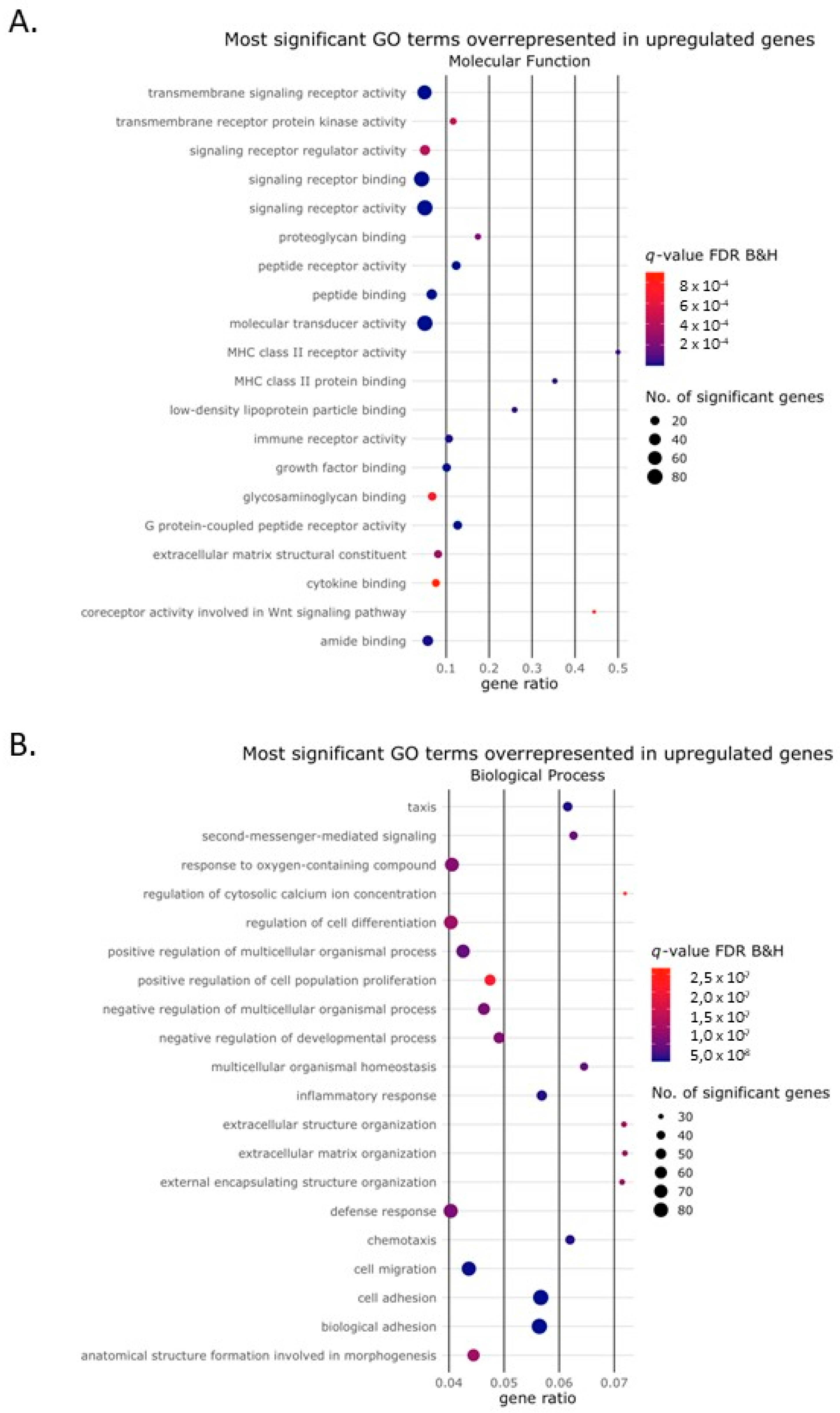
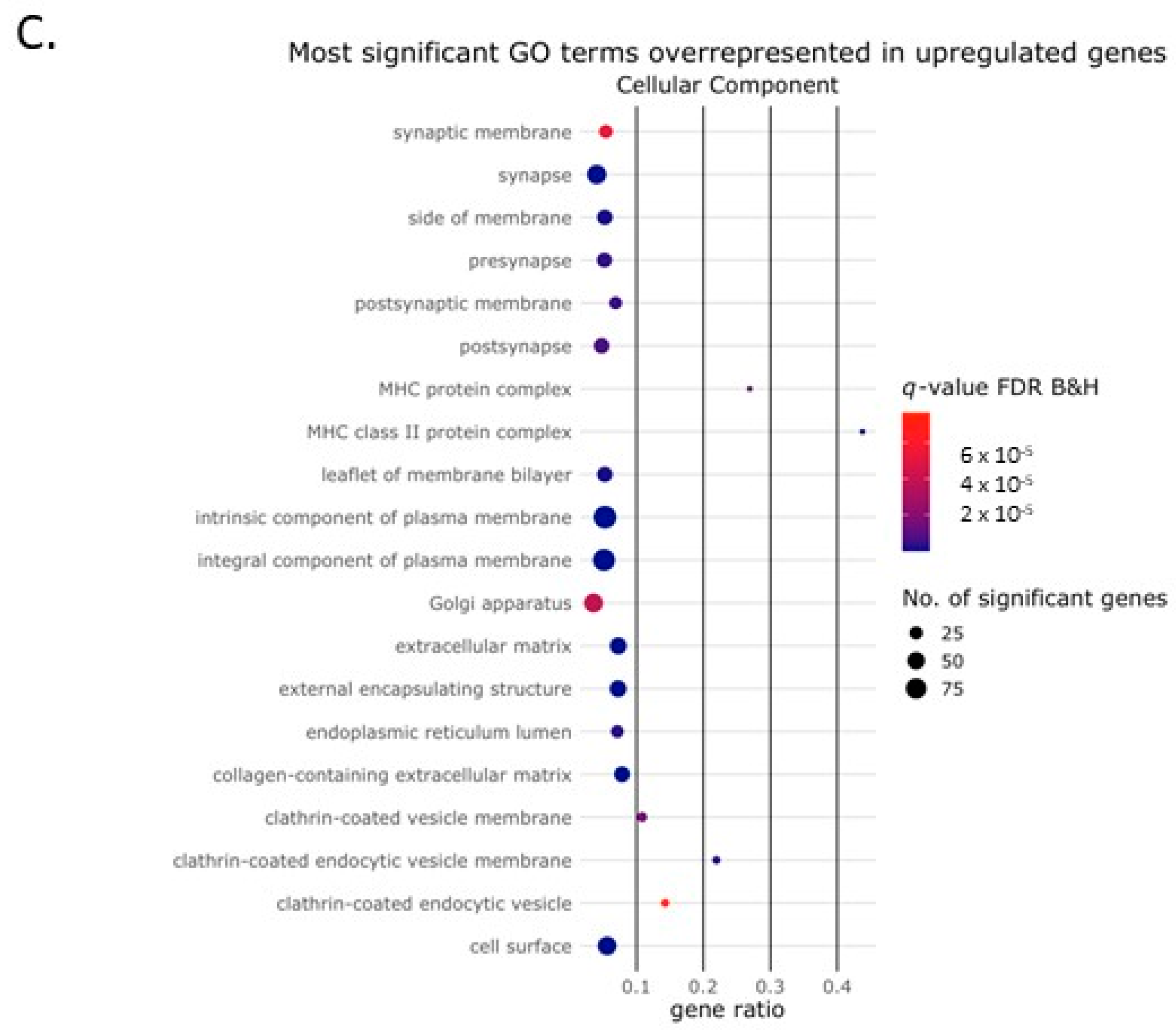
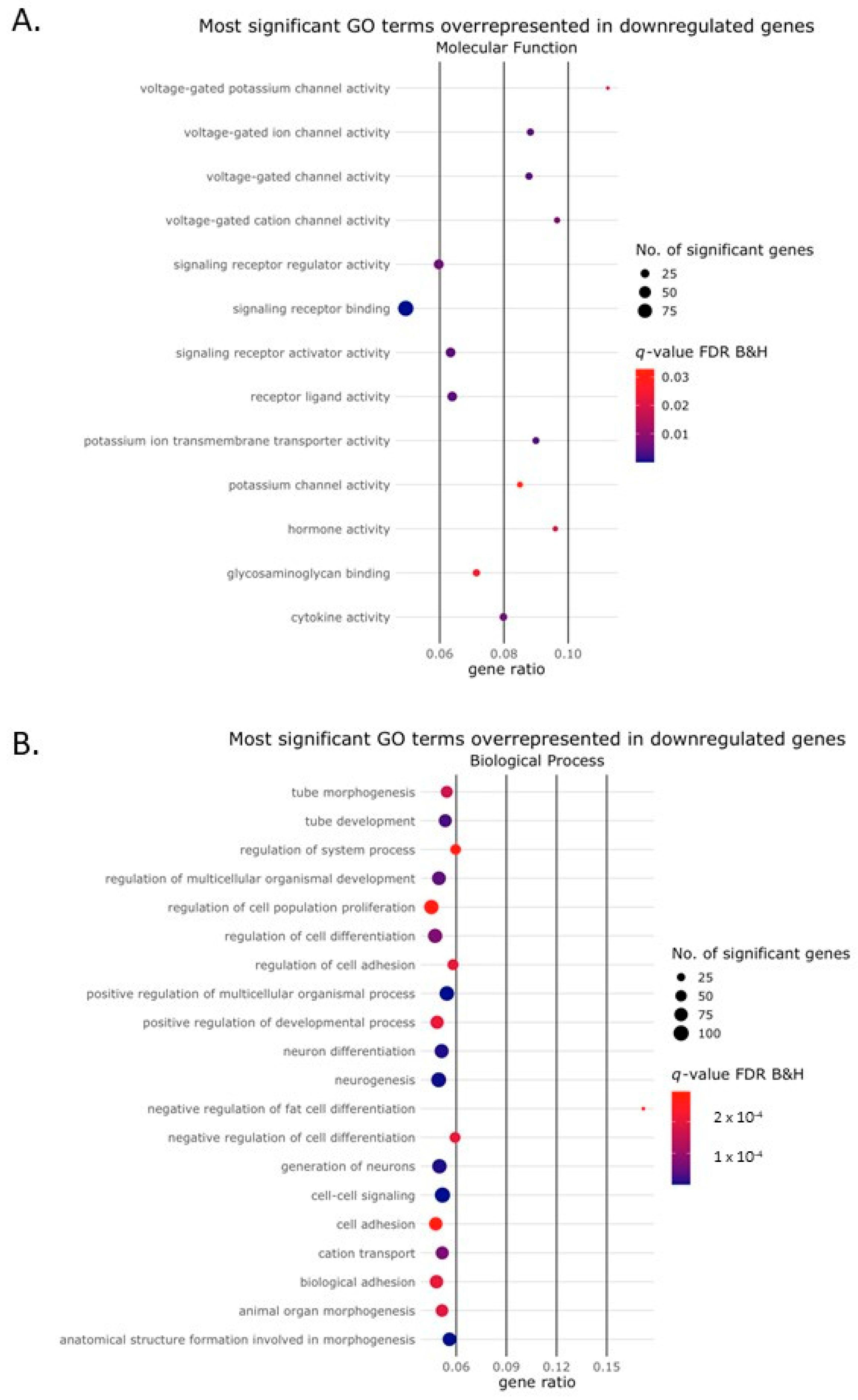
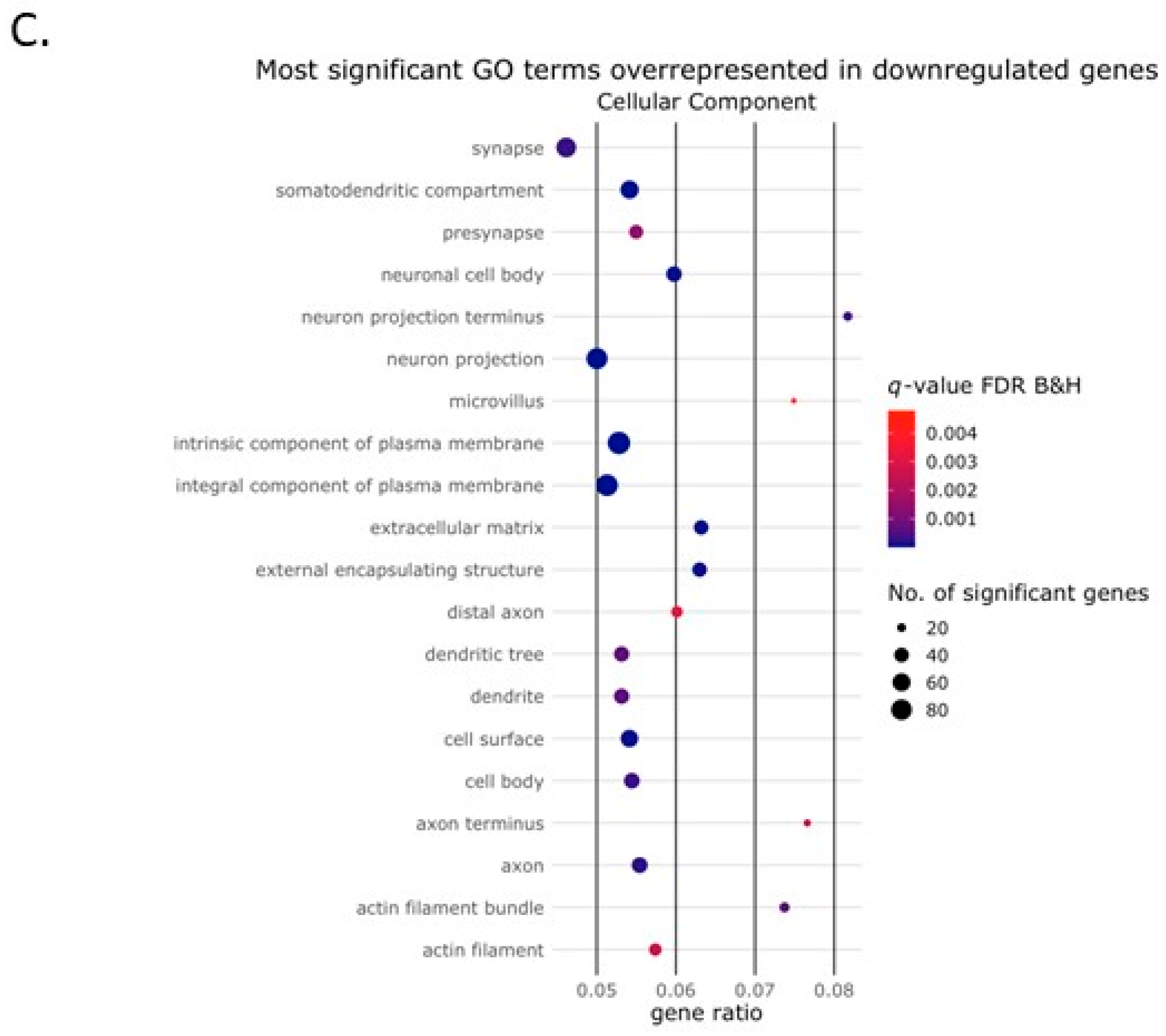
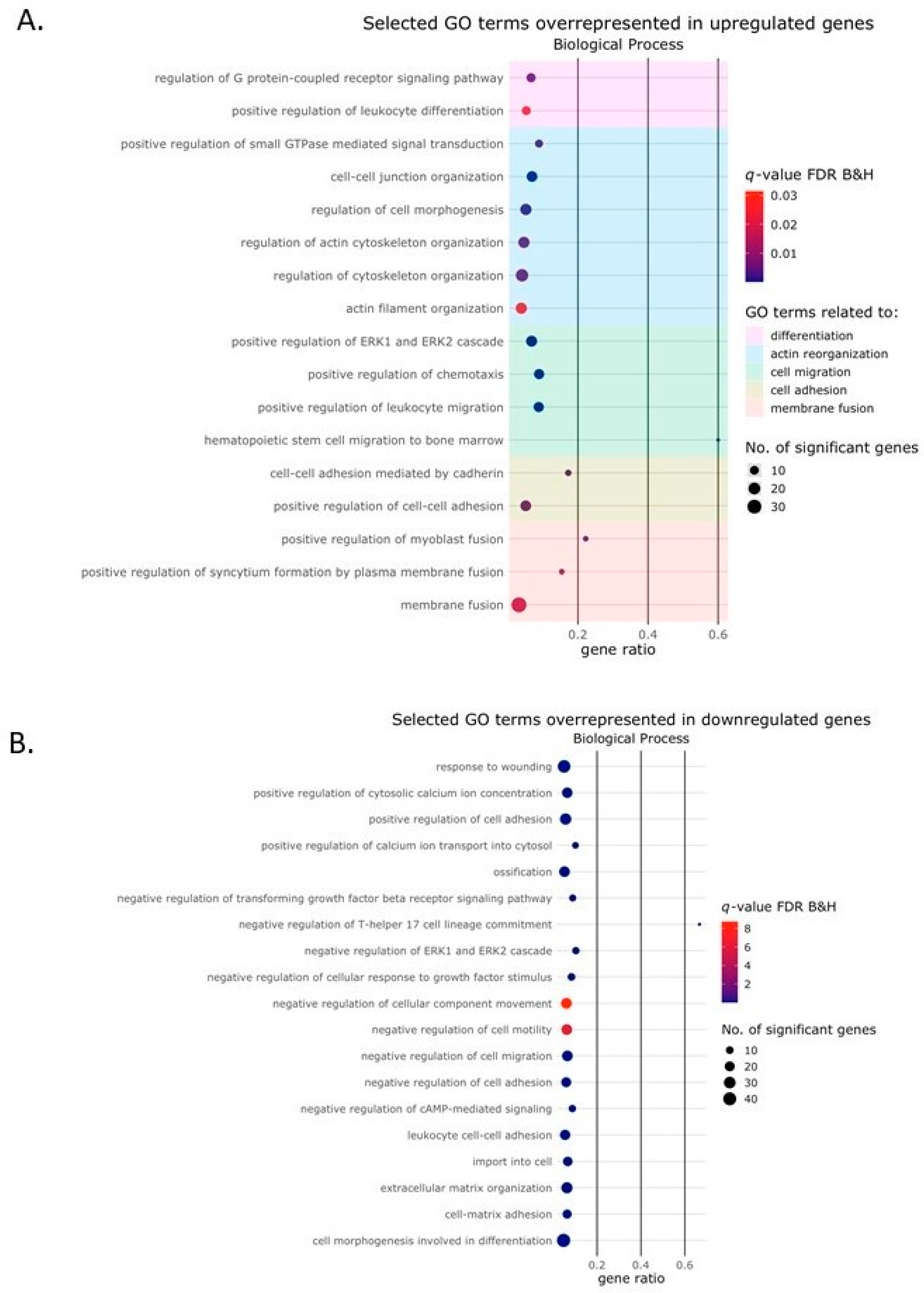
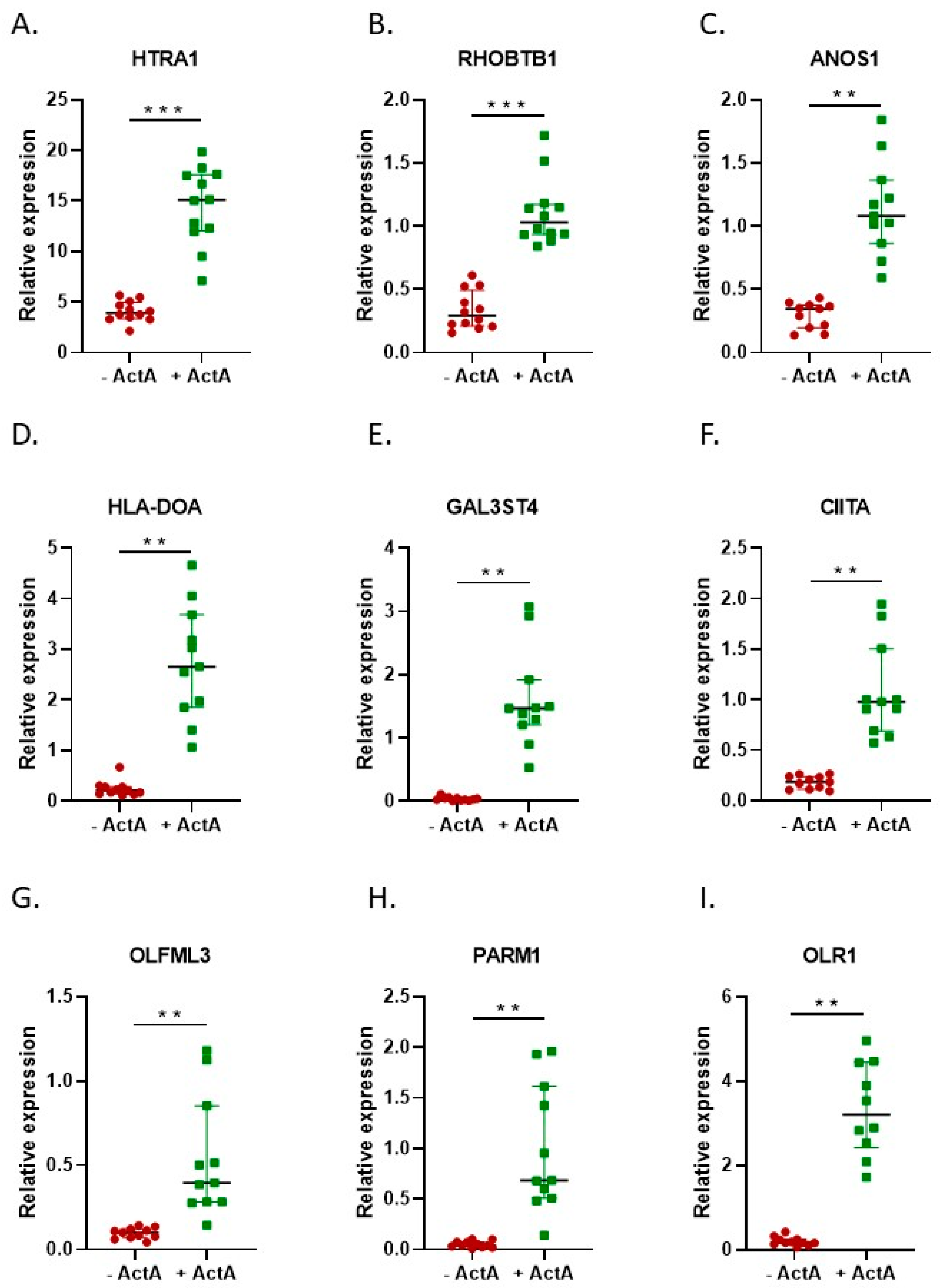
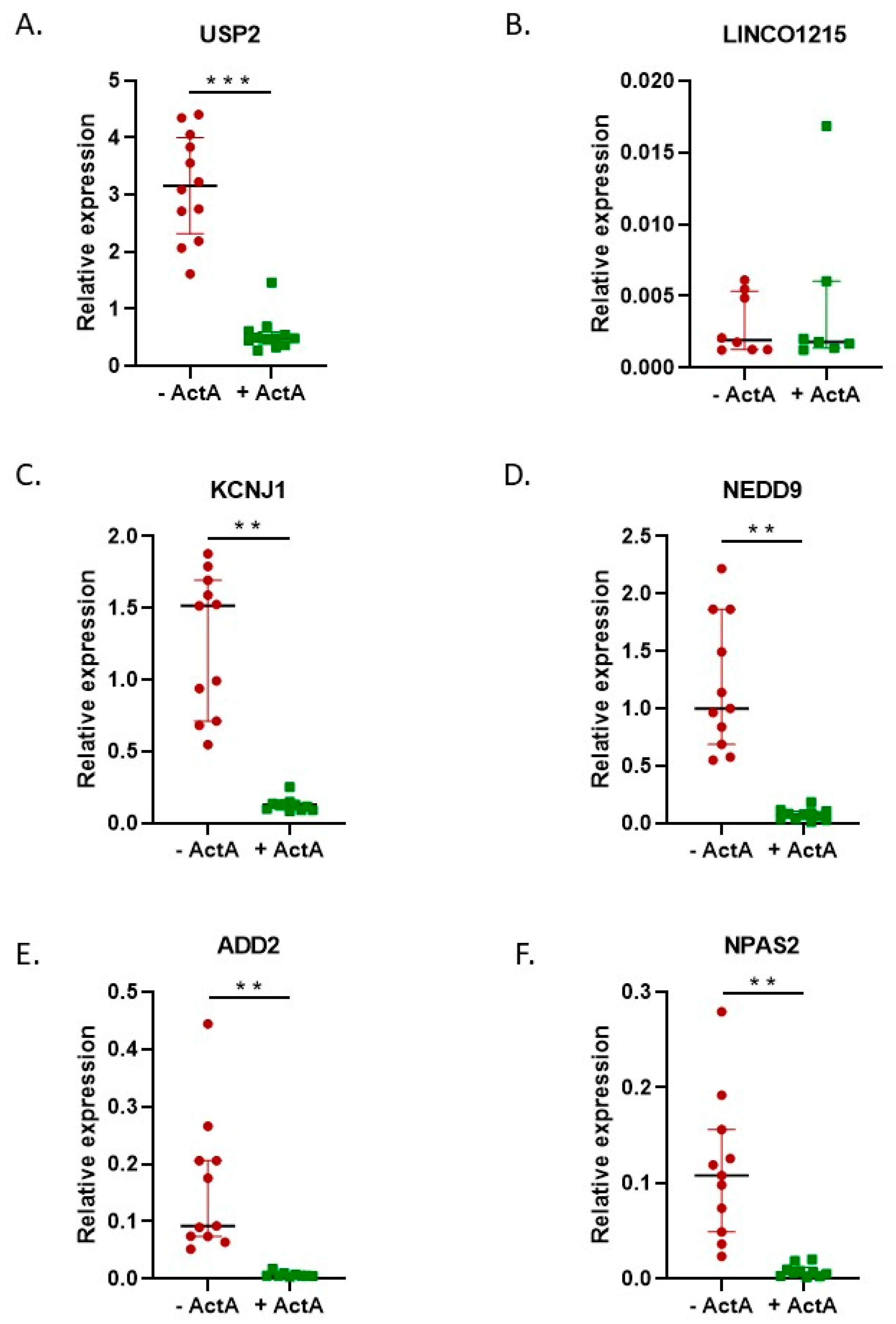
2.2. Activin-A Activates Osteoclast-Related Pathways
2.3. Activin-A-Induced Differentially Expressed Genes Can Be Linked to Cell Fusion
3. Discussion
4. Material and Methods
4.1. Osteoclastogenesis and RNA Isolation
4.2. RNA Sequencing
4.3. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
4.4. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Le Merrer, M.; Glaser, D.L.; Pignolo, R.J.; Goldsby, R.E.; Kitterman, J.A.; Groppe, J.; Shore, E.M. Fibrodysplasia ossificans progressiva. Best Pract. Res. Clin. Rheumatol. 2008, 22, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Tabas, J.A.; Gannon, F.H.; Finkel, G.; Hahn, G.V.; Zasloff, M.A. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J. Bone Jt. Surg. Am. Vol. 1993, 75, 220–230. [Google Scholar] [CrossRef]
- Olsen, O.E.; Wader, K.F.; Hella, H.; Mylin, A.K.; Turesson, I.; Nesthus, I.; Waage, A.; Sundan, A.; Holien, T. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signal. CCS 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [Green Version]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Bio. Med. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Irie, N. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 2008, 473, 201–209. [Google Scholar] [CrossRef]
- Sorensen, M.G.; Henriksen, K.; Schaller, S.; Henriksen, D.B.; Nielsen, F.C.; Dziegiel, M.H.; Karsdal, M.A. Characterization of osteoclasts derived from CD14+ monocytes isolated from peripheral blood. J. Bone Miner. Metab. 2007, 25, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Osdoby, P.; Martini, M.C.; Caplan, A.I. Isolated osteoclasts and their presumed progenitor cells, the monocyte, in culture. J. Exp. Zool. 1982, 224, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Piper, K.; Boyde, A.; Jones, S.J. The relationship between the number of nuclei of an osteoclast and its resorptive capability in vitro. Anat. Embryol. 1992, 186, 291–299. [Google Scholar] [CrossRef]
- Schoenmaker, T.; Botman, E.; Sariyildiz, M.; Micha, D.; Netelenbos, C.; Bravenboer, N.; Kelder, A.; Eekhoff, E.M.W.; De Vries, T.J. Activin-A Induces Fewer, but Larger Osteoclasts From Monocytes in Both Healthy Controls and Fibrodysplasia Ossificans Progressiva Patients. Front. Endocrinol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Pereira, M.; Petretto, E.; Gordon, S.; Bassett, J.H.D.; Williams, G.R.; Behmoaras, J. Common signalling pathways in macrophage and osteoclast multinucleation. J. Cell Sci. 2018, 131, jcs216267. [Google Scholar] [CrossRef] [Green Version]
- Hernández, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, N.; Nakachi, Y.; Yokoo, T.; Ichihara, T.; Eriksson, T.; Yonemoto, Y.; Kato, T.; Ogata, H.; Fujimoto, N.; Kobayashi, Y.; et al. Murine osteoclasts secrete serine protease HtrA1 capable of degrading osteoprotegerin in the bone microenvironment. Commun. Biol. 2019, 2, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benasciutti, E.; Mariani, E.; Oliva, L.; Scolari, M.; Perilli, E.; Barras, E.; Milan, E.; Orfanelli, U.; Fazzalari, N.L.; Campana, L.; et al. MHC class II transactivator is an in vivo regulator of osteoclast differentiation and bone homeostasis co-opted from adaptive immunity. J. Bone Miner. Res.: Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Mukai, M.; Suruga, N.; Saeki, N.; Ogawa, K. EphA receptors and ephrin-A ligands are upregulated by monocytic differentiation/maturation and promote cell adhesion and protrusion formation in HL60 monocytes. BMC Cell Biol. 2017, 18, 28. [Google Scholar] [CrossRef] [Green Version]
- Aspenström, P.; Fransson, A.; Saras, J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef]
- Wright, L.M.; Maloney, W.; Yu, X.; Kindle, L.; Collin-Osdoby, P.; Osdoby, P. Stromal cell-derived factor-1 binding to its chemokine receptor CXCR4 on precursor cells promotes the chemotactic recruitment, development and survival of human osteoclasts. Bone 2005, 36, 840–853. [Google Scholar] [CrossRef]
- Ishii, M.; Kikuta, J.; Shimazu, Y.; Meier-Schellersheim, M.; Germain, R.N. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J. Exp. Med. 2010, 207, 2793–2798. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.Y.; Choi, H.; Neff, L.; Wu, Y.; Saito, H.; Ferguson, S.M.; De Camilli, P.; Baron, R. Dynamin and endocytosis are required for the fusion of osteoclasts and myoblasts. J. Cell Biol. 2014, 207, 73–89. [Google Scholar] [CrossRef]
- Omata, Y.; Nakamura, S.; Koyama, T.; Yasui, T.; Hirose, J.; Izawa, N.; Matsumoto, T.; Imai, Y.; Seo, S.; Kurokawa, M.; et al. Identification of Nedd9 as a TGF-β-Smad2/3 Target Gene Involved in RANKL-Induced Osteoclastogenesis by Comprehensive Analysis. PLoS ONE 2016, 11, e0157992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. Elife 2020, 9, e54582. [Google Scholar] [CrossRef] [PubMed]
- Kajita, T.; Ariyoshi, W.; Okinaga, T.; Mitsugi, S.; Tominaga, K.; Nishihara, T. Mechanisms involved in enhancement of osteoclast formation by activin-A. J. Cell. Biochem. 2018, 119, 6974–6985. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, T.; Mokry, M.; Micha, D.; Netelenbos, C.; Bravenboer, N.; Gilijamse, M.; Eekhoff, E.M.W.; de Vries, T.J. Activin-A Induces Early Differential Gene Expression Exclusively in Periodontal Ligament Fibroblasts from Fibrodysplasia Ossificans Progressiva Patients. Biomedicines 2021, 9, 629. [Google Scholar] [CrossRef]
- Mundy, C.; Yao, L.; Sinha, S.; Chung, J.; Rux, D.; Catheline, S.E.; Koyama, E.; Qin, L.; Pacifici, M. Activin A promotes the development of acquired heterotopic ossification and is an effective target for disease attenuation in mice. Sci. Signal. 2021, 14, eabd0536. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell. Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Kang, D.H.; Choi, M.; Chang, S.; Lee, M.Y.; Lee, D.J.; Choi, K.; Park, J.; Han, E.C.; Hwang, D.; Kwon, K.; et al. Vascular Proteomics Reveal Novel Proteins Involved in SMC Phenotypic Change: OLR1 as a SMC Receptor Regulating Proliferation and Inflammatory Response. PLoS ONE 2015, 10, e0133845. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ding, Z.; Lin, J.; Guo, Z.; Mehta, J.L. LOX-1 in macrophage migration in response to ox-LDL and the involvement of calpains. Biochem. Biophys. Res. Commun. 2015, 467, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Kitano, V.J.F.; Ohyama, Y.; Hayashida, C.; Ito, J.; Okayasu, M.; Sato, T.; Ogasawara, T.; Tsujita, M.; Kakino, A.; Shimada, J.; et al. LDL uptake-dependent phosphatidylethanolamine translocation to the cell surface promotes fusion of osteoclast-like cells. J. Cell Sci. 2020, 133, jcs243840. [Google Scholar] [CrossRef] [PubMed]
- Nakayachi, M.; Ito, J.; Hayashida, C.; Ohyama, Y.; Kakino, A.; Okayasu, M.; Sato, T.; Ogasawara, T.; Kaneda, T.; Suda, N.; et al. Lectin-like oxidized low-density lipoprotein receptor-1 abrogation causes resistance to inflammatory bone destruction in mice, despite promoting osteoclastogenesis in the steady state. Bone 2015, 75, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef]
- Taylor, R.M.; Kashima, T.G.; Knowles, H.J.; Athanasou, N.A. VEGF, FLT3 ligand, PlGF and HGF can substitute for M-CSF to induce human osteoclast formation: Implications for giant cell tumour pathobiology. Lab. Invest. 2012, 92, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Gu, J.; Liu, X.; Zhu, J.; Wang, Q.; Gao, Q.; Zhang, J.; Cheng, L.; Tong, X.; Qi, X.; et al. Inhibition of osteoclast bone resorption activity through osteoprotegerin-induced damage of the sealing zone. Int. J. Mol. Med. 2014, 34, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Sumida, G.M.; Yamada, S. Self-contact elimination by membrane fusion. Proc. Natl. Acad. Sci. USA 2013, 110, 18958–18963. [Google Scholar] [CrossRef] [Green Version]
- Mensah, K.A.; Ritchlin, C.T.; Schwarz, E.M. RANKL induces heterogeneous DC-STAMP(lo) and DC-STAMP(hi) osteoclast precursors of which the DC-STAMP(lo) precursors are the master fusogens. J. Cell. Physiol. 2010, 223, 76–83. [Google Scholar]
- Galliano, M.F.; Huet, C.; Frygelius, J.; Polgren, A.; Wewer, U.M.; Engvall, E. Binding of ADAM12, a marker of skeletal muscle regeneration, to the muscle-specific actin-binding protein, alpha -actinin-2, is required for myoblast fusion. J. Biol. Chem. 2000, 275, 13933–13939. [Google Scholar] [CrossRef] [Green Version]
- Hochreiter-Hufford, A.E.; Lee, C.S.; Kinchen, J.M.; Sokolowski, J.D.; Arandjelovic, S.; Call, J.A.; Klibanov, A.L.; Yan, Z.; Mandell, J.W.; Ravichandran, K.S. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 2013, 497, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Verrier, S.; Hogan, A.; McKie, N.; Horton, M. ADAM gene expression and regulation during human osteoclast formation. Bone 2004, 35, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Rumpler, M.; Würger, T.; Roschger, P.; Zwettler, E.; Sturmlechner, I.; Altmann, P.; Fratzl, P.; Rogers, M.J.; Klaushofer, K. Osteoclasts on bone and dentin in vitro: Mechanism of trail formation and comparison of resorption behavior. Calcif. Tissue Int. 2013, 93, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insogna, K.; Tanaka, S.; Neff, L.; Horne, W.; Levy, J.; Baron, R. Role of c-Src in cellular events associated with colony-stimulating factor-1-induced spreading in osteoclasts. Mol. Reprod. Dev. 1997, 46, 104–108. [Google Scholar] [CrossRef]
- Stanley, A.; Tichy, E.D.; Kocan, J.; Roberts, D.W.; Shore, E.M.; Mourkioti, F. Dynamics of skeletal muscle-resident stem cells during myogenesis in fibrodysplasia ossificans progressiva. NPJ Regen. Med. 2022, 7, 5. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.L.; Mansell, A.; Patella, S.; Scott, B.J.; Hedger, M.P.; de Kretser, D.M.; Phillips, D.J. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc. Natl. Acad. Sci. USA 2007, 104, 16239–16244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.L.; Brauman, J.N.; Groome, N.P.; de Kretser, D.M.; Phillips, D.J. Activin A release into the circulation is an early event in systemic inflammation and precedes the release of follistatin. Endocrinology 2000, 141, 1905–1908. [Google Scholar] [CrossRef]
- Reith, W.; LeibundGut-Landmann, S.; Waldburger, J.M. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 2005, 5, 793–806. [Google Scholar] [CrossRef]
- Madel, M.B.; Ibáñez, L.; Wakkach, A.; de Vries, T.J.; Teti, A.; Apparailly, F.; Blin-Wakkach, C. Immune Function and Diversity of Osteoclasts in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 1408. [Google Scholar] [CrossRef]
- Li, H.; Hong, S.; Qian, J.; Zheng, Y.; Yang, J.; Yi, Q. Cross talk between the bone and immune systems: Osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 2010, 116, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, J.; Deichsel, A.; Stange, K.; Ast, C.; Bocciardi, R.; Ravazzolo, R.; Di Rocco, M.; Ferrari, P.; Landi, A.; Kaplan, F.S.; et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum. Mol. Genet. 2014, 23, 5364–5377. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego Romero, I.; Pai, A.A.; Tung, J.; Gilad, Y. RNA-seq: Impact of RNA degradation on transcript quantification. BMC Biol. 2014, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Sex | Age |
|---|---|---|
| Control 1 | Female | 27 |
| FOP 1 | Female | 26 |
| Control 2 | Male | 23 |
| FOP 2 | Male | 19 |
| Control 3 | Female | 24 |
| FOP 3 | Female | 24 |
| Control 4 | Female | 29 |
| FOP 4 | Female | 30 |
| Control 5 | Female | 25 |
| FOP 5 | Female | 22 |
| Control 6 | Male | 69 |
| FOP 6 | Male | 67 |
| Genes | Oligonucleotide Sequence, 5′–3′ | Amplicon Length (bp) | Ensembl ID |
|---|---|---|---|
| PBGD | TGCAGTTTGAAATCATTGCTATGTC | 84 | ENSG00000256269 |
| AACAGGCTTTTCTCTCCAATCTTAGA | |||
| HTRA1 | ATCAAGGATGTGGATGAGAAAGC | 61 | ENSG00000166033 |
| GCTTGCCCTGGTGGTCAAT | |||
| RHOBTB1 | CAGTGTATGCTCCAAGTTCCGTAA | 75 | ENSG00000072422 |
| CGGTGCCGCTCGAAGTATT | |||
| USP2 | GGGAACACGTGCTTCATGAA | 64 | ENSG00000036672 |
| AATCTCTCAACTCCCGAGTGTTG | |||
| ANOS1 | CAGTGGCCCAGACCACAGA | 59 | ENSG00000011201 |
| CCATCGGCTGGGTCTTATGT | |||
| LINC01215 | AGAATGCACCTATTGGCTCACA | 66 | ENSG00000271856 |
| CTGCATTGTTATCATCACGACTTTC | |||
| HLA-DOA | CAATCAAAGCCCATCTGGACAT | 137 | ENSG00000204252 |
| GTCCACGATGCAGATGAGGAT | |||
| GAL3ST4 | TCCTCTGTCACCACATGAGGTT | 142 | ENSG00000197093 |
| AAGGCTGATGAGGTGGATTTATAGTAG | |||
| KCNJ1 | TTCGGAAATGGGTCGTCACT | 62 | ENSG00000151704 |
| GGAGACTAGCCTTGCTCTTTGC | |||
| NEDD9 | CTGGATGGATGACTACGATTACGT | 68 | ENSG00000111859 |
| GCTCTTTCTGTTGCCTCTCAAAC | |||
| CIITA | GCTCTACTCAGAACCCGACACA | 63 | ENSG00000179583 |
| TCACACAACAGCCTGCTGAAC | |||
| ADD2 | CCCAAGACCACGTGGATGA | 75 | ENSG00000075340 |
| TGGGTTTTCGATGCGAATC | |||
| OLFML3 | GGTGACAGACTGTGGCTACACAA | 66 | ENSG00000116774 |
| CCACCAAATCGCTTCAGAATC | |||
| PARM1 | CGTGGTGCTGCTGGTGTTT | 61 | ENSG00000169116 |
| TCCATAGGAGGAATGCCTGATT | |||
| NPAS2 | GGCAGCATCATCTATGTCTCTGA | 67 | ENSG00000170485 |
| CCATGACATCCGACGGTAAAT | |||
| OLR1 | CCAGCCTGATGAGAAGTCAAATG | 72 | ENSG00000173391 |
| AGGCACCACCATGGAGAGTAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoenmaker, T.; Zwaak, J.; Loos, B.G.; Volckmann, R.; Koster, J.; Eekhoff, E.M.W.; de Vries, T.J. Transcriptomic Differences Underlying the Activin-A Induced Large Osteoclast Formation in Both Healthy Control and Fibrodysplasia Ossificans Progressiva Osteoclasts. Int. J. Mol. Sci. 2023, 24, 6822. https://doi.org/10.3390/ijms24076822
Schoenmaker T, Zwaak J, Loos BG, Volckmann R, Koster J, Eekhoff EMW, de Vries TJ. Transcriptomic Differences Underlying the Activin-A Induced Large Osteoclast Formation in Both Healthy Control and Fibrodysplasia Ossificans Progressiva Osteoclasts. International Journal of Molecular Sciences. 2023; 24(7):6822. https://doi.org/10.3390/ijms24076822
Chicago/Turabian StyleSchoenmaker, Ton, Joy Zwaak, Bruno G. Loos, Richard Volckmann, Jan Koster, E. Marelise W. Eekhoff, and Teun J. de Vries. 2023. "Transcriptomic Differences Underlying the Activin-A Induced Large Osteoclast Formation in Both Healthy Control and Fibrodysplasia Ossificans Progressiva Osteoclasts" International Journal of Molecular Sciences 24, no. 7: 6822. https://doi.org/10.3390/ijms24076822