2.1. Illustrative Crystal Geometry of Cesium Tetrel Halide Perovskites
The crystal structures of CsGeX
3 (X = Cl, Br, I) have been known for some time [
45]. The crystal structure of the high-temperature
Pmm phase of CsPbI
3 was reported in 2008 [
46], while the low-temperature orthorhombic structures
Pmnb (ICSD ref: 27979),
Pbnm (ICSD: 434338; 21955) [
47],
Pnam (ICSD ref: 264725) [
48], and
Pnma (ICSD ref: 32301–32314; 161480; 29350; 27484; 20759; 17016) [
49] of the same system have been known since 1959. Regardless of the nature of the temperature phase, the TtX
64− framework is common, featuring corner-sharing octahedra. These are tilted in the low-temperature phases along the crystallographic axes but linearly arranged in the high-temperature cubic phase [
6,
50,
51]. These features are evident in the orthorhombic, tetragonal, and cubic phases of CsSnI
3 (see
Figure 1a–c, respectively). They are also reminiscent of cesium lead halide perovskites, CsPbI
3. The high-to-low temperature phases of CsPbI
3 [
52] are called the
α (645K),
β (510K), and
γ (325K) phases, with space groups
Pmm,
P4/mbm, and
Pbnm, respectively. A common feature of these systems is that the TtX
3− anions are linked to each other through tetrel bonds, a feature revealed in this study, and these bonds are responsible for the formation of the 3D inorganic frameworks. All these perovskite systems mentioned above are semiconductors, and several of them have found application in photovoltaics. However, the black orthorhombic form of CsSnI
3 features p-type metallic behavior with low carrier density, despite an optical band gap of 1.3 eV [
53].
- (a)
Cesium Tetrel Iodide Perovskites
The unit-cell (top) and cage-like (bottom) structures of cesium tetrel iodide perovskites, CsTtI
3 (Tt = Pb, Sn and Ge), are shown in
Figure 2a–c. The unit cell, an ion pair, [Cs
+•TtI
3−], when periodically expanded, reveals a cage-like structure. The formation of the latter is driven by Tt···I tetrel bonds, which are equivalent and linear when Tt = Pb and Sn and quasi-linear and non-equivalent when Tt = Ge. For instance, the distances associated with the Tt–I coordinate bonds within the [TtI
3−] fragment and Tt···I tetrel bonds between a [TtI
3−] pair are 3.145 and 3.110 Å in cubic (
Pm-m) CsPbI
3 (
Figure 2a) and CsSnI
3 (
Figure 2b), respectively. Because of the small size of the Ge cation, CsGeI
3 perovskite is not cubic (
Figure 2c) and crystalizes in the rhombohedral space group
R3m. Each of the three equivalent Ge–I coordinate bonds within the [GeI
3−] fragment is 2.747 Å, and each of the three equivalent Ge···I tetrel bonds between four [GeI
3−] fragments linked with each other in the CsGeI
3 is 3.262 Å. The latter are directional but quasi-linear, ∠I–Ge···I = 169.3°, which is equivalent to the tilting angle, ∠Ge···I–Ge = 169.3°, of the octahedra along each of the three principal axes. There is no structure of CsSiI
3 deposited in the ICSD.
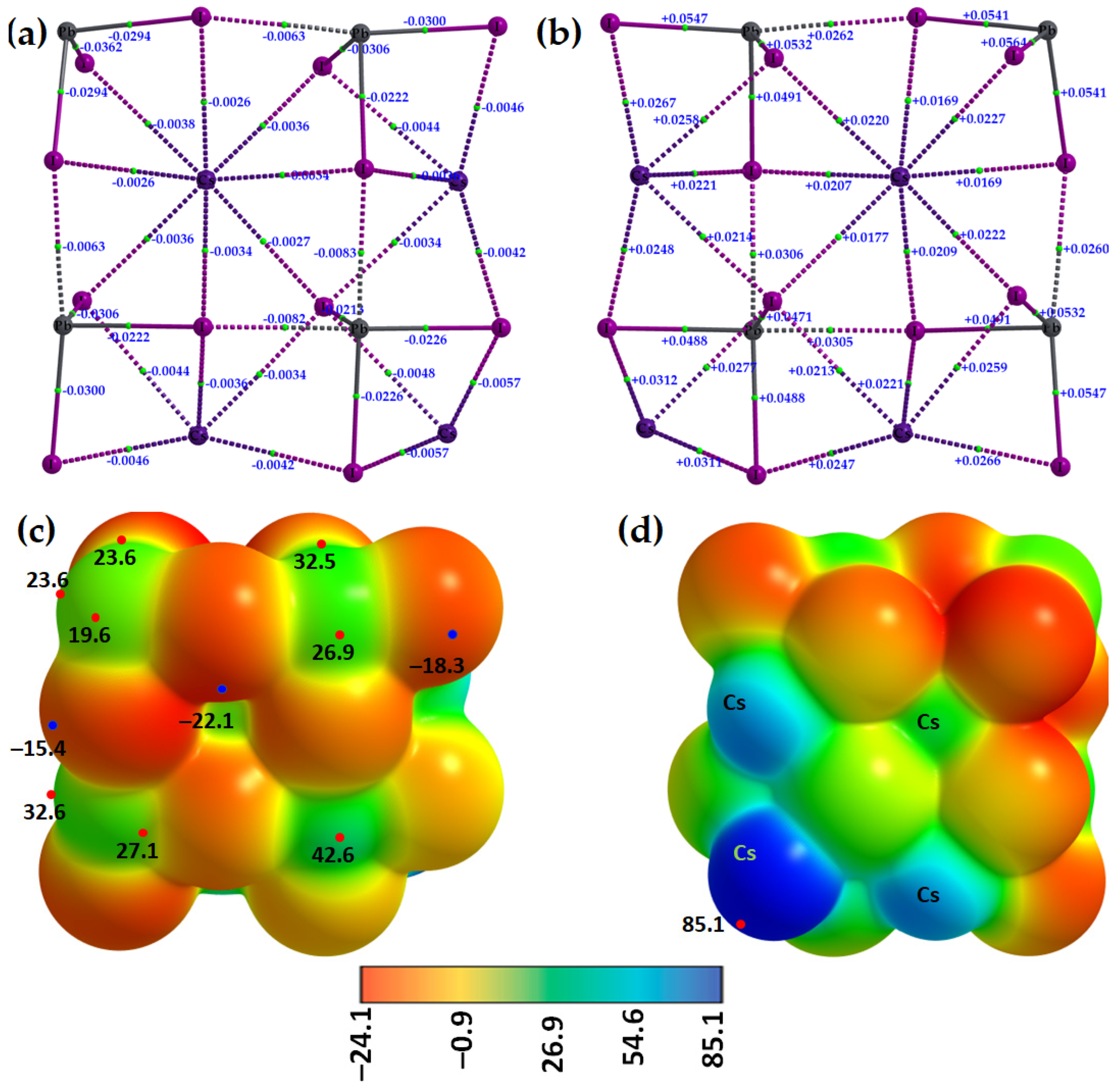
In order to shed some light on the why the entirely negative anions [TtI
3−] attract each other, thus forming tetrel bonds between them (
Figure 2, bottom), we performed both QTAIM and MESP analyses. The molecular graphs of cesium tetrel iodide perovskite ion pairs, [Cs
+•TtI
3−] (Tt = Si, Ge, Sn and Pb), are shown in
Figure 3a–d. The topology of the bond paths suggests that Tt–I coordinate bonds, characterized by solid lines, are shorter and stronger than the Cs···I close contacts, described by dotted lines; this is in line with the charge density values at their corresponding bcps, meaning that the Cs···I close contacts are weaker than the Tt–I coordinate bonds. Focusing on CsPbI
3 as a representative example of this series, each PbI
3− unit, which is a face of the PbI
64− octahedron, is involved in an attractive coulombic interaction with Cs
+, thereby forming the ion pair [Cs
+•PbI
3−]. As shown in
Figure 2a (bottom), a single cation simultaneously interacts with eight PbI
3− faces of eight PbI
64− octahedra, forming a cage-like structure, with Cs
+ trapped inside the cage formed by the eight PbI
64− octahedra. In other words, the arrangement between the anion and the cation in CsPbI
3 is such that each face of the corner-shared PbI
64− octahedra hosts a Cs
+ cation (cf.
Figure 2a).
The question that immediately arises as to why the cesium tetrel halide perovskites with Tt = Pb, Sn, and Ge have been synthesized, but not with Tt = Si. This is answered below. In short, it is the result of the nucleophilic nature of the electrostatic potential on the surface of Si in molecular [Cs+•SiI3−] that prevents self-assembly of these recurring units.
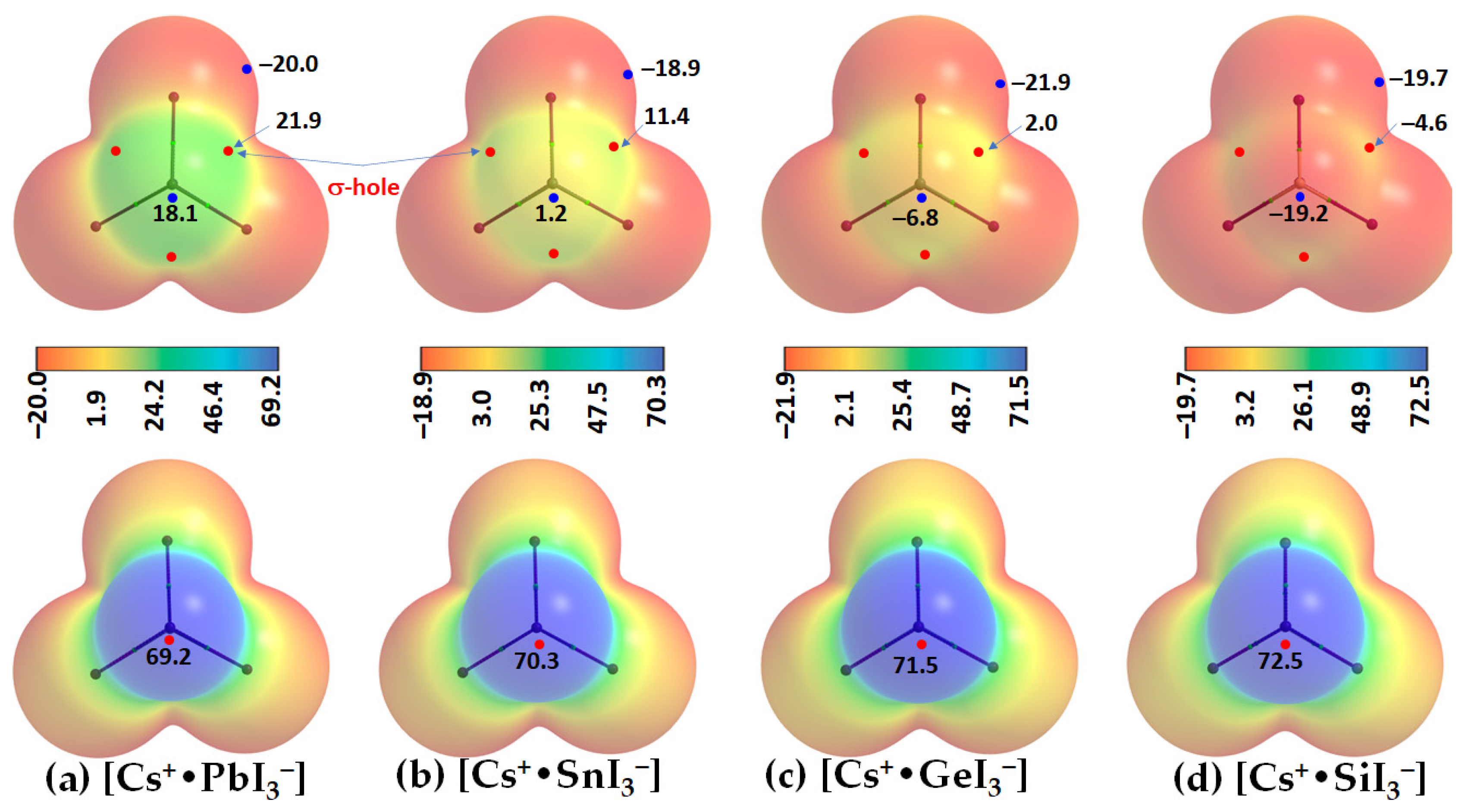
Two distinct features can be readily seen from
Figure 4. First, the tetrel atom in the ion pairs carries either a negative or a positive potential (
VS,max < 0 or
VS,max > 0) along the outermost extension of each I–Tt bond. In specific,
VS,max > 0 in [Cs
+•PbI
3−], [Cs
+•SnI
3−], and [Cs
+•GeI
3−], becomes progressively less positive, and switches to
VS,max < 0 in [Cs
+•SiI
3−]. This is the result of the polarizing field induced by Cs
+ when it is placed close to the [TtI
3−] anion. The polarization of the surface charge density of Tt in [TtI
3−] is accompanied by an appreciable transfer of charge from the iodide to Cs
+ cation, varying between 0.085 and 0.116
e. This causes the charge density on the surface of Tt to be rearranged in such a manner as to produce depleted regions of charge density on its surface. The charge density-depleted regions appear along the outermost extensions of the I–Tt bonds when Tt = Pb, Sn, or Ge, but not when Tt = Si. These charge density regions on the tetrel atoms along the extension of the three I–Tt bonds are characteristic of σ-holes since they appear opposite to the I–Tt σ coordinate bonds; thus, σ-holes on Tt are electrophilic in [Cs
+•PbI
3−], [Cs
+•SnI
3−], and [Cs
+•GeI
3−] (
VS,max > 0; see
Figure 4a–c) but nucleophilic in [Cs
+•SnI
3−] (
VS,max < 0;
Figure 4d).
Secondly, a local most minimum of potential,
VS,min, is found on the Tt atom in each [Cs
+•TtI
3−], appearing on its surface along the outer extension of the
C3v axis. It is positive in [Cs
+•PbI
3−] and [Cs
+•SnI
3−] and negative in [Cs
+•GeI
3−] and [Cs
+•SnI
3−]. The strength of the minimum of potential decreases in the order Pb (
VS,min = 18.1 kcal mol
−1) < Sn (1.2 kcal mol
−1) < Ge (–6.8 kcal mol
−1) < Si (–19.2 kcal mol
−1). This indicates that Pb and Sn are entirely electrophilic, unlike Ge and Si. The MESP graphs also suggest that the stereochemically active lone pair of the Tt sites are squeezed onto the surfaces of the iodides when Tt = Pb and Sn, but not when Tt = Ge and Si. This conclusion is in accordance with a previous study where it was suggested that “a stereochemically active lone pair of electrons of the Pb atom may lie between the two I atoms in the plane” [
55]. It was recently argued that the s
2 lone pair on heavy main-group elements in their lower oxidation states is responsible for the emergence of polar ground states in some ferroic materials and causes a crystallographically hidden, locally distorted state that appears upon warming, a phenomenon referred to as emphanisis [
56]. Others have argued that PbO and PbS in both the rocksalt and litharge structures, which have distorted Pb
2+ octahedra, are not the result of chemically inert, stereochemically active lone pairs, but instead are the result of asymmetric electron densities that rely on direct electronic interaction with the coordinated anions [
57]. Further discussion on the importance of stereochemically active lone pairs on Pb in Pb(II) halide compounds can be found elsewhere [
58].
The local most maximum potential on the surface of Cs
+ in the ion pairs is positive,
VS,max > 0, and Cs
+ becomes increasingly more electrophilic down the series [Cs
+•PbI
3−], [Cs
+•SnI
3−], [Cs
+•GeI
3−], and [Cs
+•SiI
3−]. This is expected, as Si is relatively more electronegative in the tetrel series, hence, the ability of Cs
+ to polarize electron density of small-sized Tt decreases. In all cases, both the lateral and axial portions of the halide atoms in [TtI
3−] are entirely negative, with
VS,min < 0, as seen in
Figure 4a–d.
Our QTAM results in
Figure 3 and
Table 1 suggest that the Si–I bonds have appreciable covalency, whereas the Tt–I (Tt = Ge, Sn, and Pb) in [Cs
+•TtI
3−] have a more ionic character. The Si–I bonds are characterized by negative values of both ∇
2ρb and
Hb at the Si–I bcps. Although the Tt–I (Tt = Ge, Sn, and Pb) bonds possess ionic character, the negative
Hb values at the Tt–I bcps are indicative of some measure of covalency. These coordinate bonds clearly have mixed bonding character. On the other hand, the cesium-centered charge-assisted alkali bonds, Cs···I, possess closed-shell character (∇
2ρb > 0 and
Hb > 0) and the charge density at the Cs···I bcps is significantly smaller than that at the Tt–I bcps.
- (b)
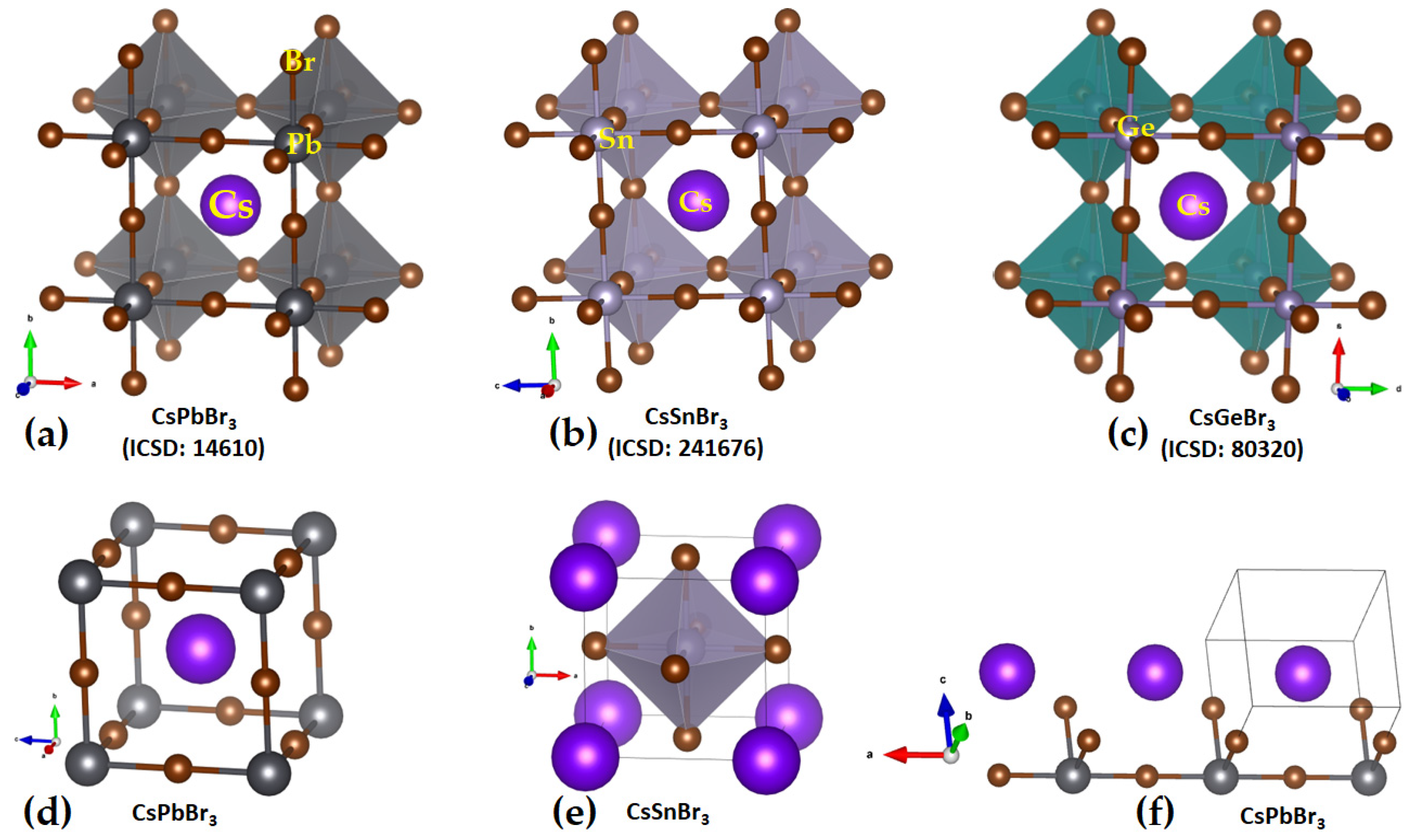
Cesium Tetrel Bromide Perovskites
The cesium tetrel bromide perovskites, [CsTtBr
3] (Tt = Pb, Sn, Ge), have been reported in different temperature crystalline phases, except for [CsSiBr
3]; the structures of the high-temperature cubic phase are shown in
Figure 5. This is probably because the surface of the Si atom along the Br–Si bond extensions is entirely negative, so the Si atom in the [Cs
+•SiBr
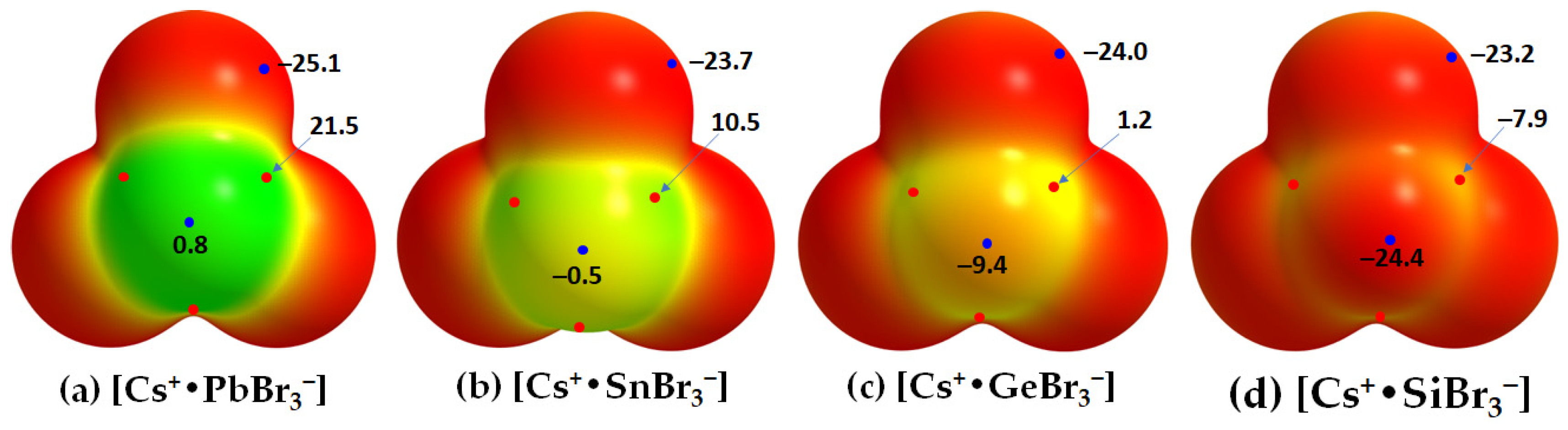
3−] ion pair is unable to coulombically attract the nucleophilic bromide in a neighboring unit. This is supported by the MESP plots of [Cs
+•TtBr
3−] shown in
Figure 6. They suggest that the surface of Pb in [Cs
+•PbBr
3−] is entirely positive along and around the outermost extension of the Br–Pb bonds (
VS,max = 21.5 kcal mol
−1 and
VS,min = 0.8 kcal mol
−1). That of Sn in [Cs
+•SnBr
3−] is appreciably positive (
VS,max = 10.5 kcal mol
−1) along and weakly negative (
VS,min = −0.5 kcal mol
−1) around the outermost extension of the Br–Sn bonds. In [Cs
+•GeBr
3−], the surface of Ge is weakly positive along and appreciably negative around the outermost extension of the Br–Ge bonds. However, in the case of [Cs
+•SiBr
3−], the
VS,max and
VS,min on Si are all negative. For all four ion pairs, both the axial and equatorial portions of the Br atom along and around the Tt–Br extensions are nucleophilic (
VS,min = −25.1 kcal mol
−1).
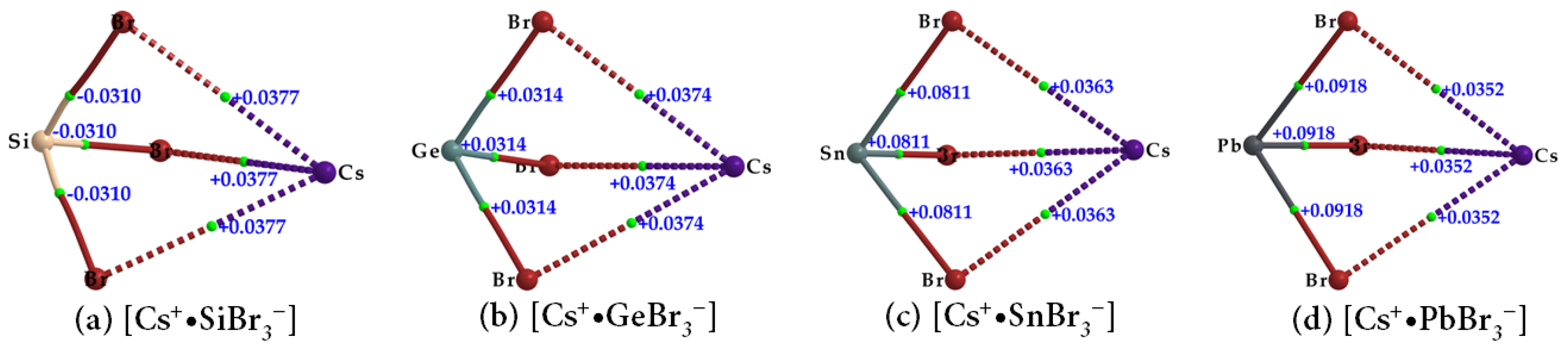
The topological charge density characteristics of the Tt–Br and Cs···Br bcps in the ion pairs [Cs
+•TtBr
3−] (Tt = Si, Ge, Sn, Pb) (
Figure 7a–d and
Table 2) were found to be very similar to those observed for the Tt–I and Cs···I bcps in [Cs
+•TtI
3−] (
Table 1). However, the charge densities at the Tt–Br and Cs···Br bcps in [Cs
+•TtBr
3−] were slightly larger; hence the strength of the Tt–Br and Cs···Br bonds are marginally stronger than the Tt–I and Cs···I bonds in [Cs
+•TtI
3−]. The values of ∇
2ρb are negative at the Si–Br bcps and positive at the Tt–Br (Tt = Ge, Sn, Pb) bcps, as seen in
Figure 7 and listed in
Table 2. The extent of charge transfer from the anion to the cation lies between 0.092 and 0.095
e.
- (c)
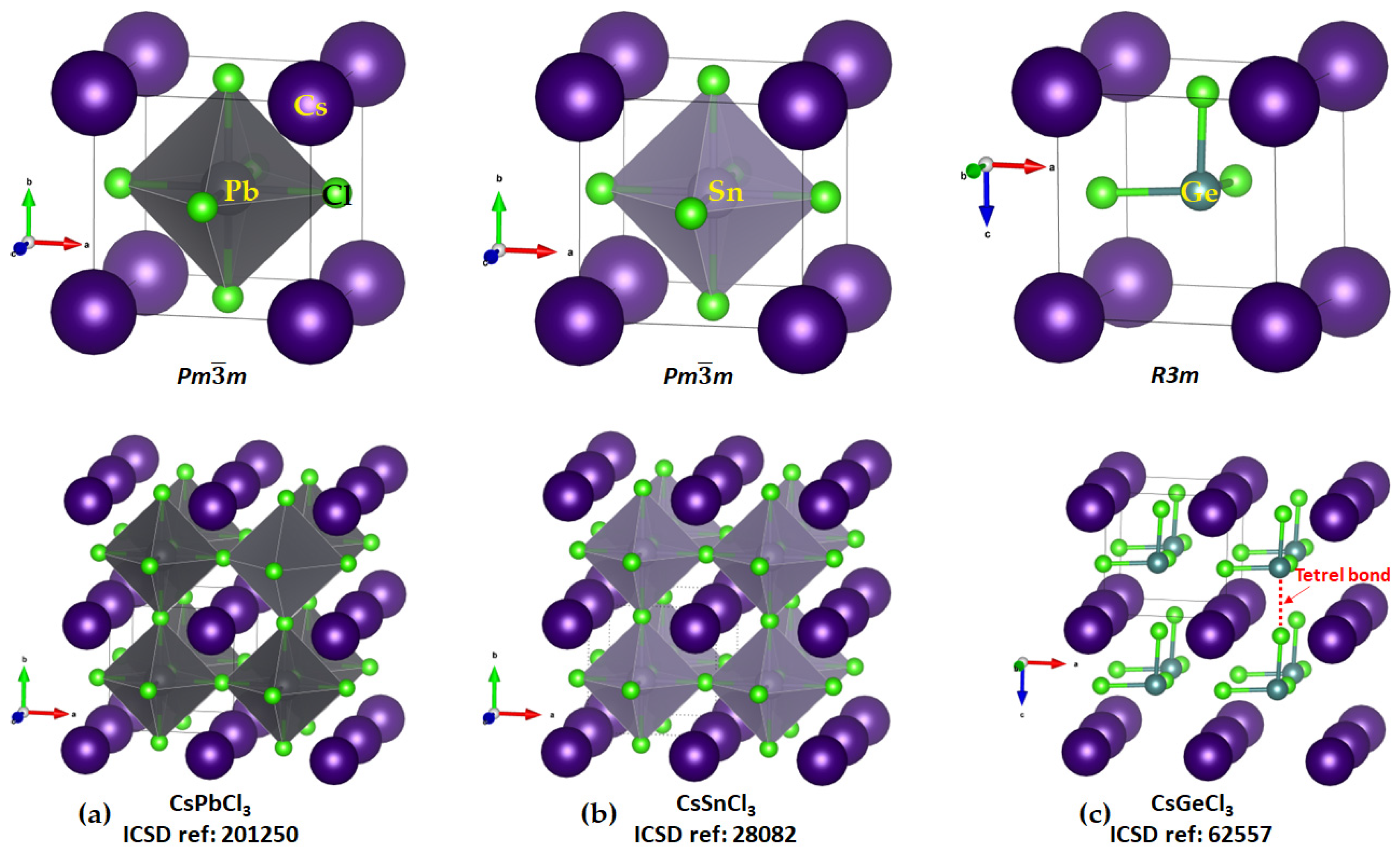
Cesium Tetrel Chloride Perovskites
The solid-state structures of CsTtCl
3 (Tt = Ge, Sn, and Pb) are known, whereas that of [CsSiCl
3] has not been reported. The connectivity between the [TtCl
3−] units that lead to the formation of the TtCl
64− octahedra in the solid state are evident in all three structures shown in
Figure 8a–c. The six Tt–Cl bonds in each polyhedron are equivalent in CsPbCl
3 and CsSiCl
3 (2.803 Å and 2.752 Å, respectively), showing that there is very little difference between the three Tt···Cl tetrel and three Tt–Cl coordinate bonds in these systems. In the case of CsGeCl
3, three of the coordinate bonds are different to the other three; the three Ge–Cl coordinate bond distances are equivalent (2.415 Å each;
Figure 8c, bottom) that are appreciably shorter than the three Ge···Cl tetrel bonds (
r(Ge···Cl) = 3.036 Å and ∠Cl–Ge···Cl = 172.1°).
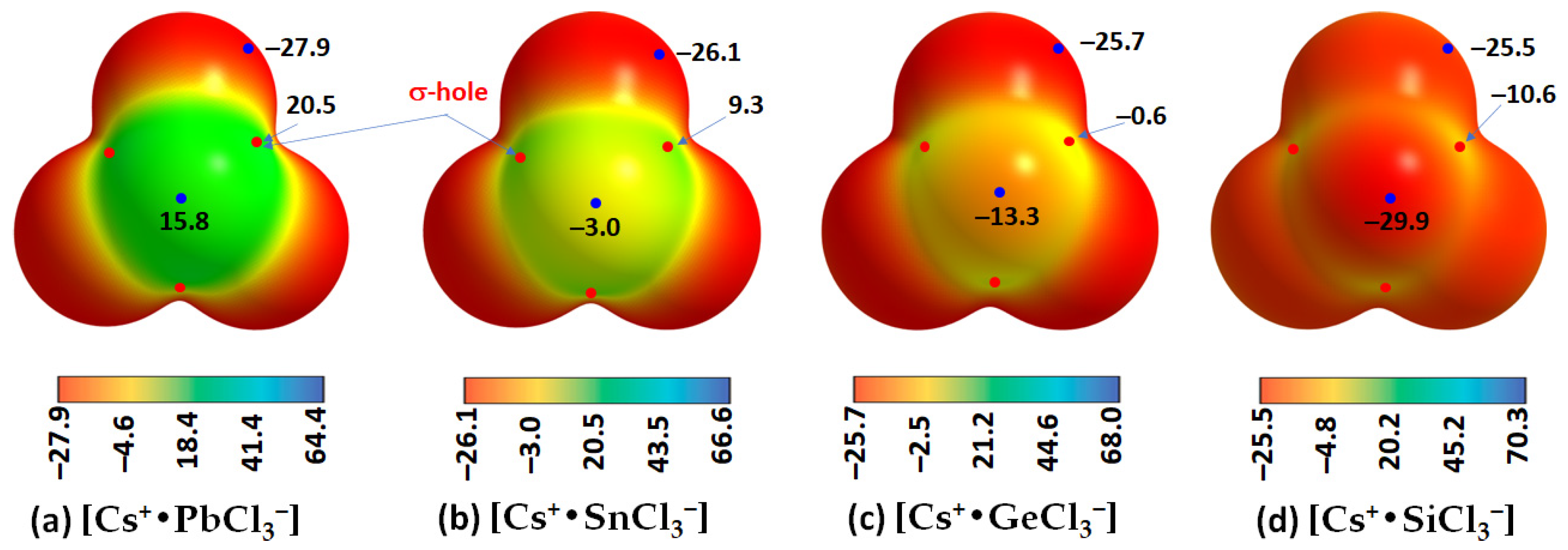
The fact that tetrel bonding plays a vital role in assembling the [TtCl
3−] units, which leads to the development of the cage-like inorganic framework, is evident from the results of the MESP analysis shown in
Figure 9a–c for the ion pairs [Cs
+•TtCl
3−]. Each of the three Cl–Tt bonds in each ion pair in [Cs
+•TtCl
3−] (Tt = Pb, Sn) contains electron density-deficient regions along the Cl–Tt bond extensions (σ-holes) with positive electrostatic potentials. However, the potential is weakly negative in [Cs
+•GeCl
3−] along the three Cl–Ge bond extensions (
VS,max = −0.6 kcal mol
−1 each,
Figure 9c) and strongly negative along the three Cl–Si bond extensions in [Cs
+•SiCl
3−] (
VS,max = −10.6 kcal mol
−1). It is clear from these results that the formation of CsTtCl
3 (Tt = Pb, Sn) perovskite systems in 3D is expected when repeating units of [Cs
+•TtCl
3−] ion pairs are in close proximity. This kind of assembly is unlikely when [Cs
+•TtCl
3−] (Tt = Ge and Si) pairs are in close proximity because of coulombic repulsion between the halogen of an ion pair in close proximity to the negative tetrel site in a neighboring unit.
We, and others, have shown on several occasions that caution needs to be exercised when the potential of a σ-hole on an atom in a molecular entity is near neutral. In such a case, a higher isoelectronic density envelope may be required for mapping the potential since the choice of isoelectronic density envelope is arbitrary. Indeed, this is the case with [Cs
+•GeCl
3−]. When the 0.001 a.u. isoelectronic density was used for mapping, the potential associated with each of the three σ-hole holes on Ge was weakly negative (
VS,max = −0.6 kcal mol
−1). However, when a 0.0015 a.u. isoelectron density was used, the
VS,max of the σ-holes on the same atom was positive,
VS,max = 4.4 kcal mol
−1. The positive nature of the σ-hole on Ge explains why Ge in [Cs
+•GeCl
3−] is capable of attracting the negative portion on the Cl atoms in a neighboring interacting ion-pair, thus leading to the formation of CsGeCl
3 perovskite crystals in the crystalline phase [
45]. By contrast, changing the value of the isodensity envelope did not change the negative character of the σ-holes on the Si atom along the Cl–Si bond extensions; thus, CsSiCl
3 structures should not be formed when the ion pairs are repeated periodically.
There is a potential maximum on the Cs atom in the ion-pair that appears along the extension of the
C3v axis. Its origin could be due to a weak Tt···Cs interaction in [Cs
+•TtCl
3−] (Tt = Si, Ge, Sn, Pb), as well as the formation of three equivalent Cs···Cl alkali bonds. The surface of Cs is strongly positive relative to that of Tt in each [Cs
+•TtCl
3−] (Tt = Si, Ge, Sn, Pb), which rationalizes why the cation lies at the center of the inorganic tetrel halide cage, thus interacting simultaneously with the lone-pair dominant regions of coordinate halides on each of the eight faces of eight octahedra (each sitting at the corner of a cage,
Figure 8).
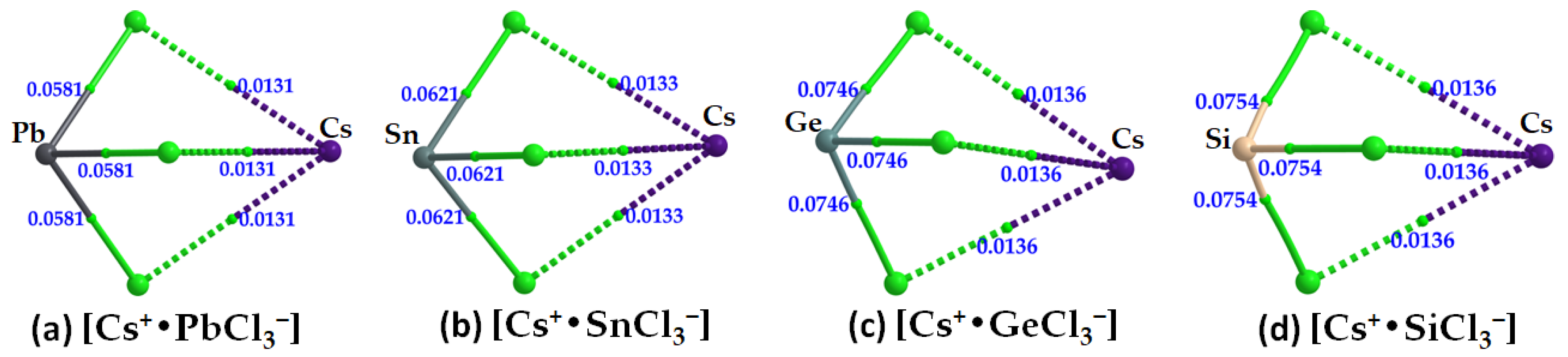
The formation of the alkali bonds in each ion pair, which is expected to mimic what occurs in the crystal (
vide infra), is evident in the molecular graphs shown in
Figure 10a–d and
Table 3. The accumulation of charge density at the Cs···Cl bcps is weaker than that at the Tt–Cl bcps. For the latter, it trends as Pb–Cl < Sn–Cl < Ge–Cl < Si–Cl, and, with ∇
2ρb > 0 and
Hb < 0 (see values in
Table 3), these bonds have mixed bonding character. This feature is clearly distinguishable from that of the alkali bonds that are largely electrostatic in character (∇
2ρb > 0 and
Hb > 0).
- (d)
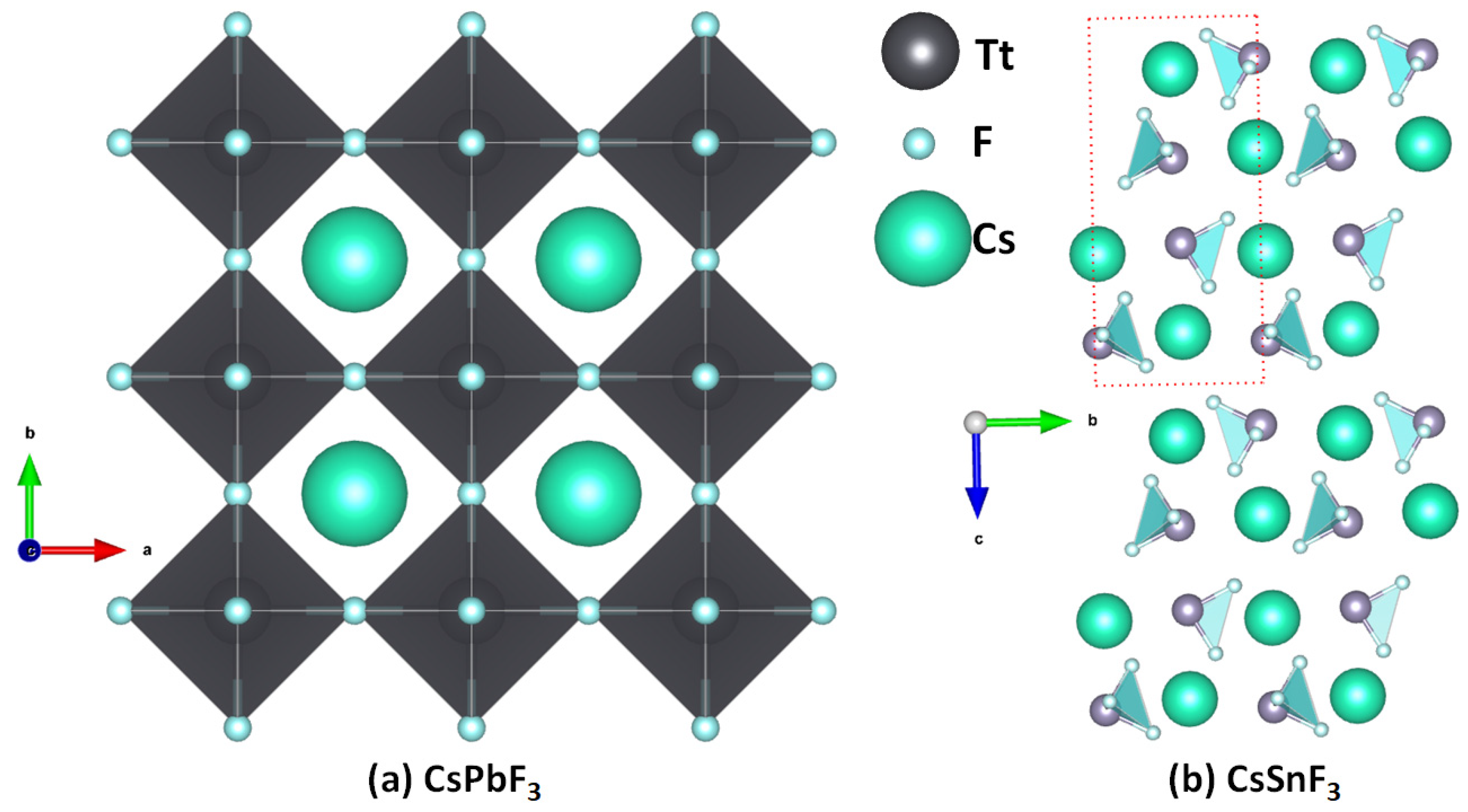
Cesium Tetrel Fluoride Perovskites
The structures of [Cs
+•TtF
3−] are similar to the other cesium halide perovskite ion pairs discussed above. While the formation of these ion-pair systems is likely in the gas phase, they are not all stable in the crystalline phase. The instability of these perovskite structures is arguably due to the mismatch between the cavity of the fluoride-based inorganic perovskite cage formed from the repeating units of [TtF
3−] and the radial size of Cs
+. This is not the case for CsPbF
3, as seen in
Figure 11a, the structure of which was reported in 1956 (cubic,
Pmm, ICSD ref: 30739 [
59]) and 2001 (ICSD refs: 93438–93439). Smith et al. [
60] have suggested that CsPbF
3 is the only experimentally synthesized AMF
3 fluoride perovskite with a polar ground state. Our search of the ICSD showed that CsSnF
3 is not cubic (space group: P12
1/n1(14) [
61]) and hence is a non-perovskite (
Figure 11b). The authors of that study suggested that this system exhibits a ‘zero-dimensional’ crystal structure with isolated SnF
3− anions separated by Cs
+ cations; again, this is not surprising since the size of the cage formed by the repeating units of the SnF
3− anion is too small to accommodate the guest Cs
+. The ICSD does not contain structures of CsTtF
3 (Tt = Ge, Sn), but it catalogues crystals such as Cs
2GeF
6 and Cs
3GeF
7, suggesting that the small size of fluoride, its low polarizability, and its high electronegativity lead it to form other types of crystal structures.
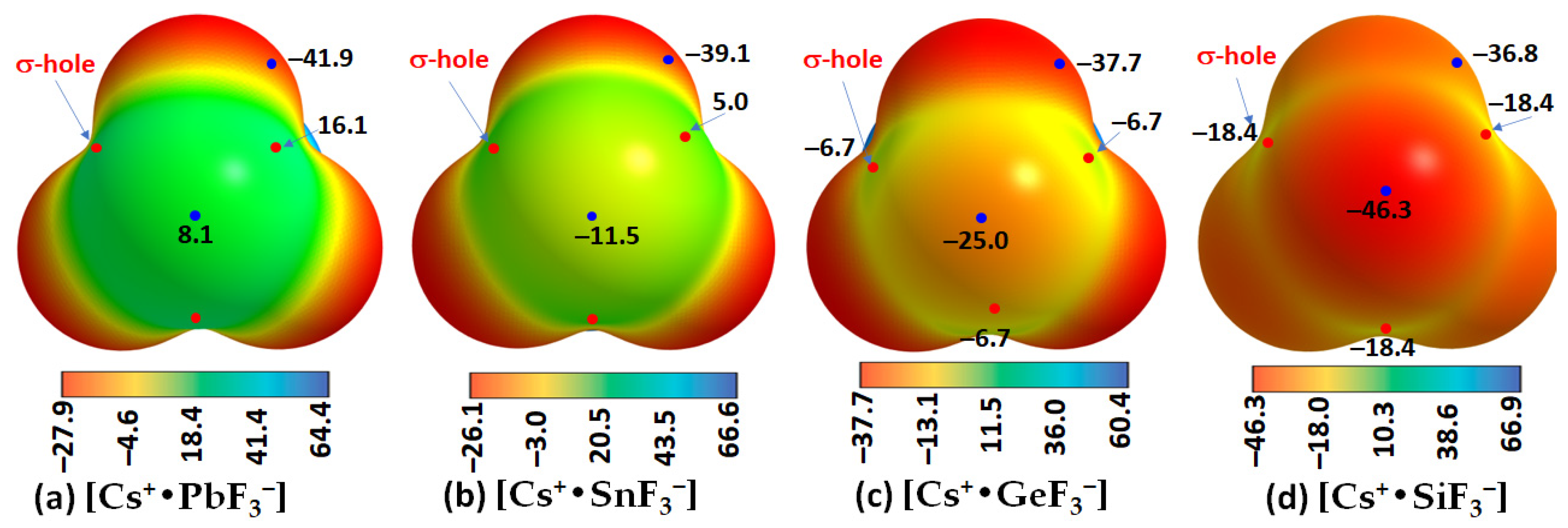
The results of our MESP calculations, shown in
Figure 12, are in accordance with these rationalizations. They suggest the feasibility of the formation of [CsTtF
3] (Tt = Pb and Sn) structures in the solid state since the surfaces of the Tt site in the [Cs
+•TtF
3−] ion-pair systems are highly electrophilic, with the former more so than the latter. Specifically, the surface of Pb in [Cs
+•PbF
3−] is entirely positive along and around the F–Pb bond extensions (
Figure 12a), whereas that of Sn is positive only along the F–Sn bond extensions, while the region around the outer extension of the
C3v axis is highly nucleophilic (
Figure 12b). These positive sites are able to engage in a coulombic attraction with the negative site on the halogen of a neighboring unit to form structures of the types shown in
Figure 11a,b, respectively. This is not the case when Tt = Ge and Si, the surfaces of which are entirely negative in [Cs
+•GeF
3−] and [Cs
+•SiF
3−] (see
Figure 12c,d), respectively.
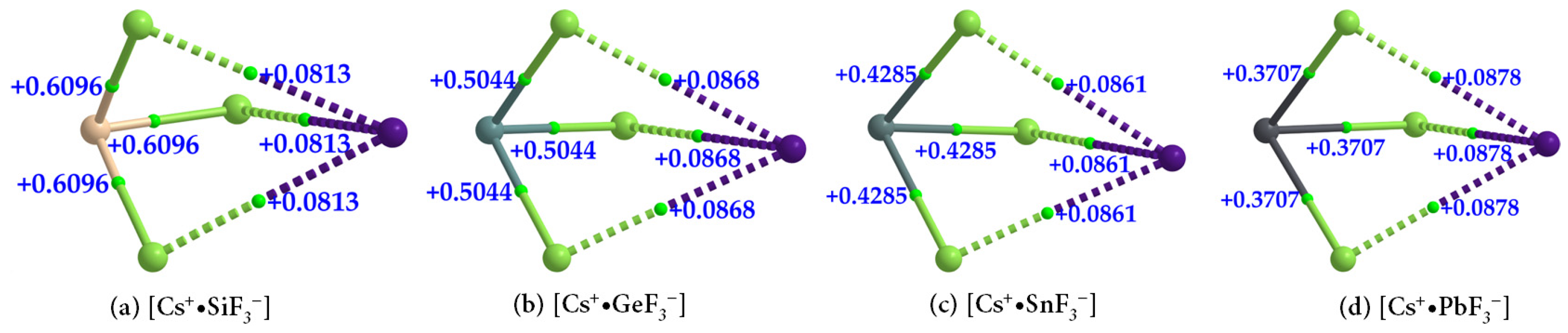
The formation of [Cs
+•TtF
3−] (Tt = Si, Ge, Sn, and Pb) ion pairs is also evident in the QTAIM-based molecular graphs shown in
Figure 13a–d. In all cases, the ∇
2ρb at the Tt–F bcps are positive, showing that they are closed-shell interactions and appreciably ionic. The values of ∇
2ρb at Tt–F bcps across the series follow the order [Cs
+•SiF
3−] > [Cs
+•GeF
3−] > [Cs
+•SnF
3−] > [Cs
+•PbF
3−], which parallels the trend in the negative
Hb values for the same bonds (
Table 4);
Hb < 0 indicates that the bonds possess some covalency. The character of these coordinate interactions deduced from ∇
2ρb and
Hb values are not the same as that found for the Cs···F bcps. For the latter, the sign of both ∇
2ρb and
Hb are positive (
Table 4), indicative of closed-shell (non-covalent) interactions.
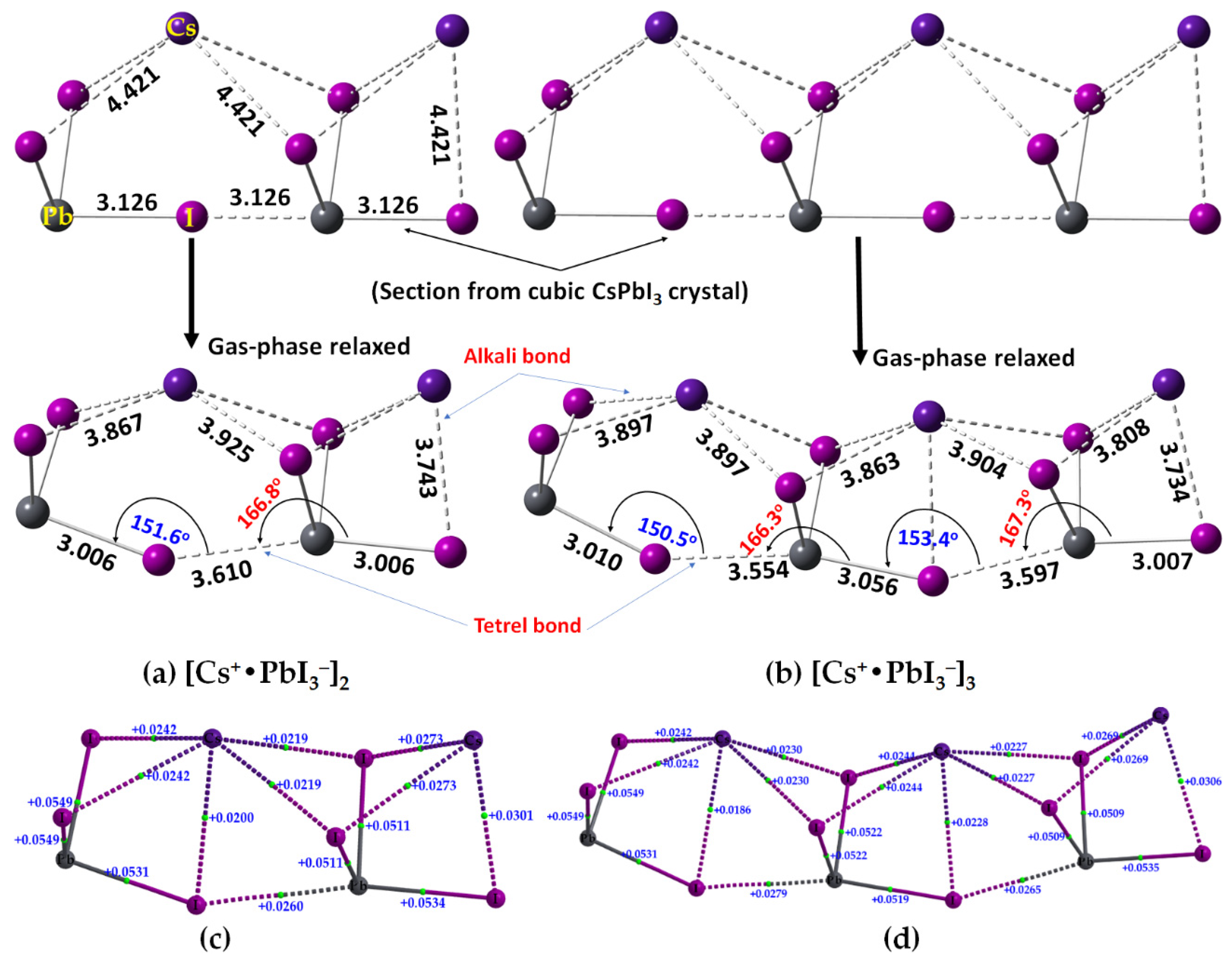
2.2. Oligomers of the [Cs+•PbI3−] Ion Pair
We have sectioned the supercell structure of cubic CsPbI
3 (
Figure 2a, bottom), and extracted the binary, trinary, and tertiary clusters in 1D. These were fully energy minimized at the same level of theory, [
ωB97XD/def2-TZVPPD]. The geometries of the resulting [Cs
+•PbI
3−]
2 dimer and [Cs
+•PbI
3−]
3 trimer are shown in
Figure 14a,b, respectively, together with their corresponding QTAIM-based molecular graphs in
Figure 14c,d, respectively. The Pb–I bonds found in the crystal (top) are significantly elongated in the gas phase (bottom) (cf.
Figure 14a,b). The ∠Pb–I···Pb angles between the ion pairs in cubic CsPbI
3 are linear, but non-linear in the gas-phase structure, leading to significant deformation passing from the solid-state structure to the gas-phase dimer and trimer. This discrepancy between the gas-phase and the solid-state geometries is not very surprising given that the role of packing forces is absent in the former. Interestingly, both the gas phase structures resemble the tilting of edge-sharing Pb–I chains in 1D, observed in the case of 3D CsPbI
3. The ∠Pb–I···Pb angles are between 150° and 154° (
Figure 14a,b, bottom), close to that seen between the edge-shared [PbI
6]
4− octahedra that are tilted relative to the corner-sharing octahedra in the low-temperature orthorhombic structure of CsPbI
3 (∠Pb–I–Pb = 148.1° along the
a-axis and 156.88° along the
c-axis; ICSD ref: 17016 [
49]). On the other hand, and as noted above, the tetrel bonds between the ion pairs are longer than the Pb–I coordinate bonds and are quasi-linear (∠I–Pb···I = 166.8° in [Cs
+•PbI
3−]
2 (
Figure 14a) and 166.3° and 167.3° in [Cs
+•PbI
3−]
3 (
Figure 14b). The physical chemistry of 1D CsPbI
3 has been experimentally investigated [
62,
63,
64]. It was shown that in the orthorhombic (
Pnma)
γ-phase, the PbI
64− octahedra tilted around all three pseudocubic axes,
a−a−c+, which is different to the tilt observed in the tetragonal (
P4/mbm),
a0a0c+,
β-phase [
65], and the bandgap increases with an increase in the octahedral tilting when the temperature cools down, allowing for the emergence of
β-CsPbI
3 and
γ-CsPbI
3 [
66].
The results of our QTAIM analysis for the [Cs
+•PbI
3−]
2 dimer (and [Cs
+•PbI
3−]
3 trimer) are given in
Table 5. The Pb–I coordinate bonds in the [Cs
+•PbI
3−]
2 dimer (and [Cs
+•PbI
3−]
3 trimer) are characterized by
ρb, ∇
2ρb and −
Hb values in the ranges of 0.0411–0.0472 (0.0379–0.0474), 0.0511–0.0116 (0.0509–0.0549), and 0.0071–0.007 (0.0058–0.0097) a.u., respectively. The corresponding values for the Cs···I alkali bonds were 0.0075–0.0116 (0.0069–0.0103), 0.0020–0.0301 (0.0186–0.0269), and −0.0008 (−0.0008) a.u., respectively. Although the former bonds possess a non-negligible amount of covalent character, the latter are purely electrostatic interactions. Their characteristics are comparable to those of I–Pb···I tetrel bonds (
ρb = 0.0139 a.u., ∇
2ρb = 0.0260 a.u., and −
Hb = 0.0001 a.u). The [Cs
+•PbI
3−]
3 trimer has two non-equivalent I–Pb···I tetrel bonds (3.554 and 3.597 Å,
Figure 14b), with
ρb = 0.0152 (0.0143) a.u., ∇
2ρb = 0.0279 (0.0265) a.u., and
Hb ≈ 0.00001 a.u for the shorter (longer) bonds. The detailed nature of ∇
2ρb at various bcps is shown in
Figure 14c,d for the [Cs
+•PbI
3−]
2 dimer and [Cs
+•PbI
3−]
3 trimer, respectively.
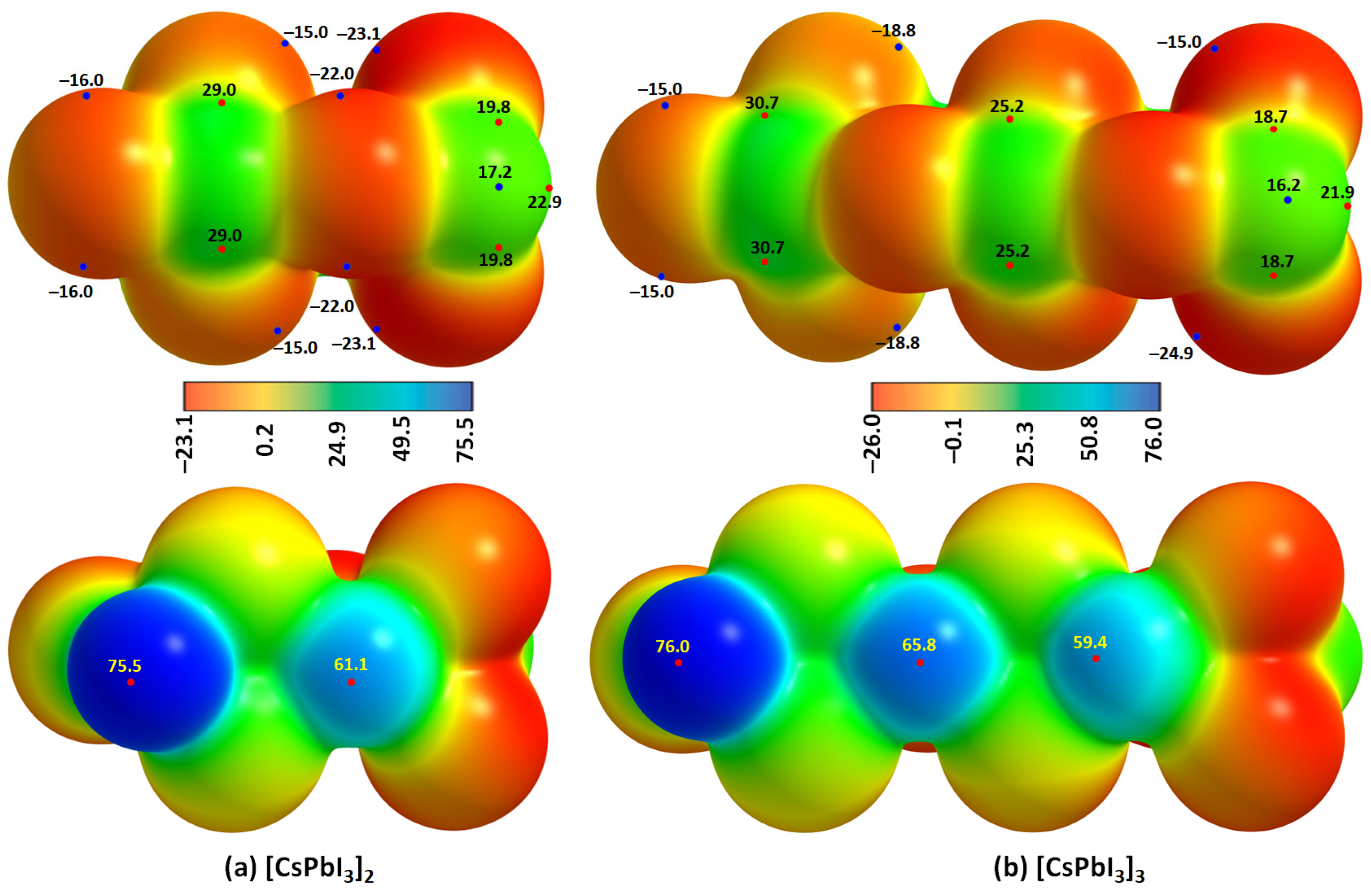
We confirmed the formation of I–Pb···I tetrel bonds in [Cs
+•PbI
3−]
2 and [Cs
+•PbI
3−]
3 oligomers using MESP results,
Figure 15a,b. This signifies that one of the positive σ-holes on the surface of Pb in the [Cs
+•PbI
3−] ion pair (
Figure 4a) is annihilated because it is engaged with the entirely negative (iodide) site in the neighboring ion pair through an electrostatic interaction, leading to the formation of an I–Pb···I tetrel bond. This causes a change in the potential minima and maxima on the surfaces of the two ion-pair entities at the equilibrium geometry of the oligomer.
An interesting feature of the [Cs
+•PbI
3−]
4 tetramer is that the Pb–I bonds are no longer equivalent, as found in cubic CsPbI
3 in 3D (
Figure 4a), but comparable with those found in the [Cs
+•PbI
3−]
2 dimer and [Cs
+•PbI
3−]
3 trimer (
vide supra). Again, this is the result of the gas phase, where the role of the periodic boundary condition is nullified and no packing forces act on the system. The [Cs
+•PbI
3−] ion pairs are free to interact with each other in the gas phase at 0 K, causing the linear Pb–I–Pb bonds found in the cubic structure of CsPbI
3 to change appreciably in a manner so as to adopt a significantly distorted geometry very similar to that observed in the low-temperature orthorhombic phase of the system (
vide supra).
The molecular graphs of two different orientations of the [Cs
+•PbI
3−]
4 tetramer are shown in
Figure 16a (with values of the potential energy density,
Vb) and
Figure 16b (∇
2ρb at the bcps). Regardless of the nature of the bonding interactions involved, the sign of
Vb is always negative and, hence, stabilizing. Values of ∇
2ρb are all positive, indicating that the bonding interactions are of the closed-shell type. While the relationship
Eb(QTAIM) = −½
Vb may be empirical, it suggests that the I–Pb···I tetrel bond is stronger than the Cs···I alkali bond (
Eb(QTAIM) values of 4.0 kcal mol
−1 vs. 1.5 kcal mol
−1).
The formation of both Pb···I tetrel bonds and Cs···I alkali bonds between four units of the [Cs
+•PbI
3−] ion pairs in the [Cs
+•PbI
3−]
4 tetramer can also be understood from the MESP plots shown in
Figure 16c,d. Upon assembly, the σ-hole on three Pb atoms in three ion pairs of the tetramer is annihilated upon its attractive engagement with the iodide atom of a neighboring ion pair, thus forming Pb···I tetrel bonds. The four tetrel centers are positive (see the four green regions in
Figure 16c), and one of them, which is not involved in the formation of the tetrel bond (
Figure 16c, top left), conceives three σ-holes on its surface; these can accept nucleophiles when in close proximity to another three ion pairs. By contrast, the Cs ions are highly electrophilic. These unequivocally provide evidence of the fact that the formation of the 3D network of the cage-like structures of cesium tetrel halide perovskites are the result of σ-hole-centered tetrel-bonded interactions between [PbI
3−] anions in the presence of Cs
+. The physical chemistry of tetrel bonds also plays a significant role in stabilizing 1D CsPbI
3, a material suitable for stable X-ray detection (sensitivity = 2.37 mC·Gy
−1·cm
−2, resistivity = 7.4 × 10
9 Ω·cm, and carrier mobility–lifetime product = 3.63 × 10
−3 cm
2·V
−1 [
62]).























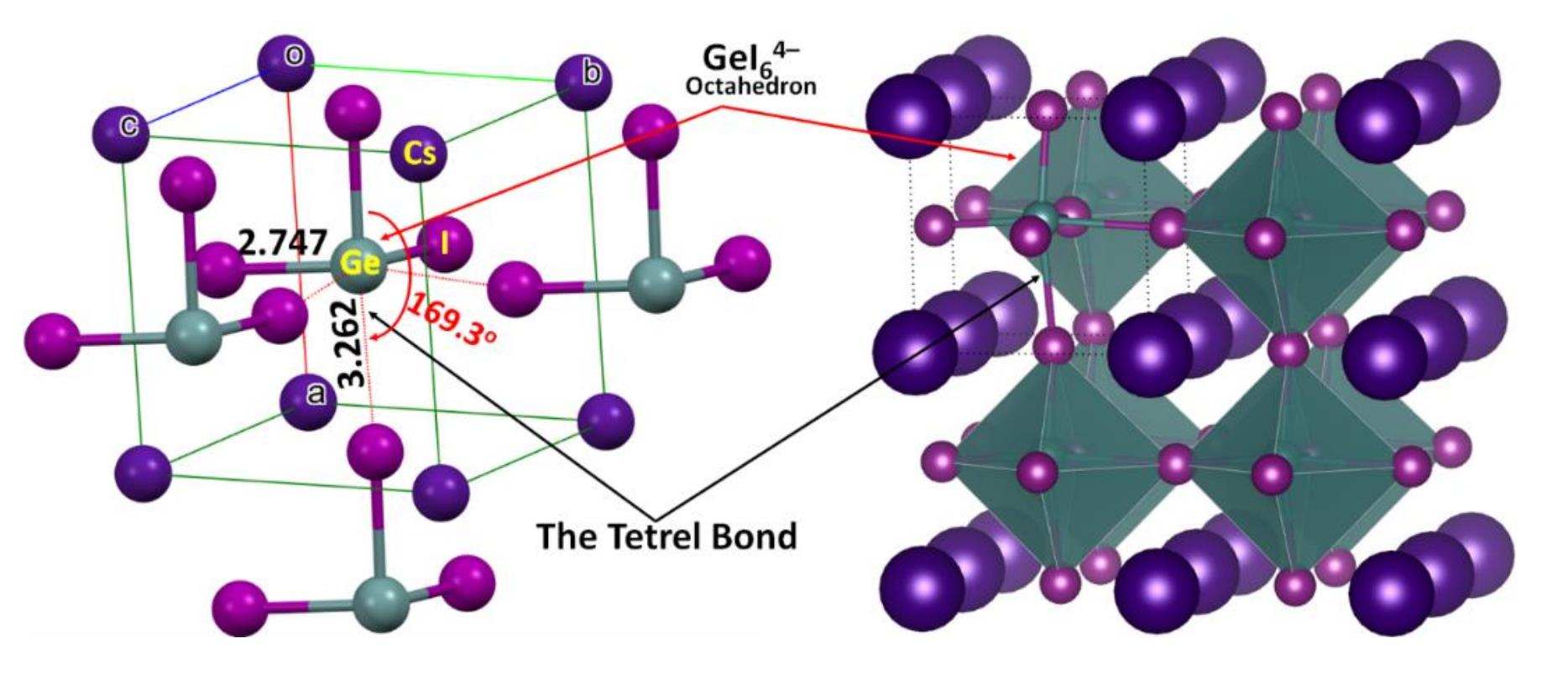{kind=link}
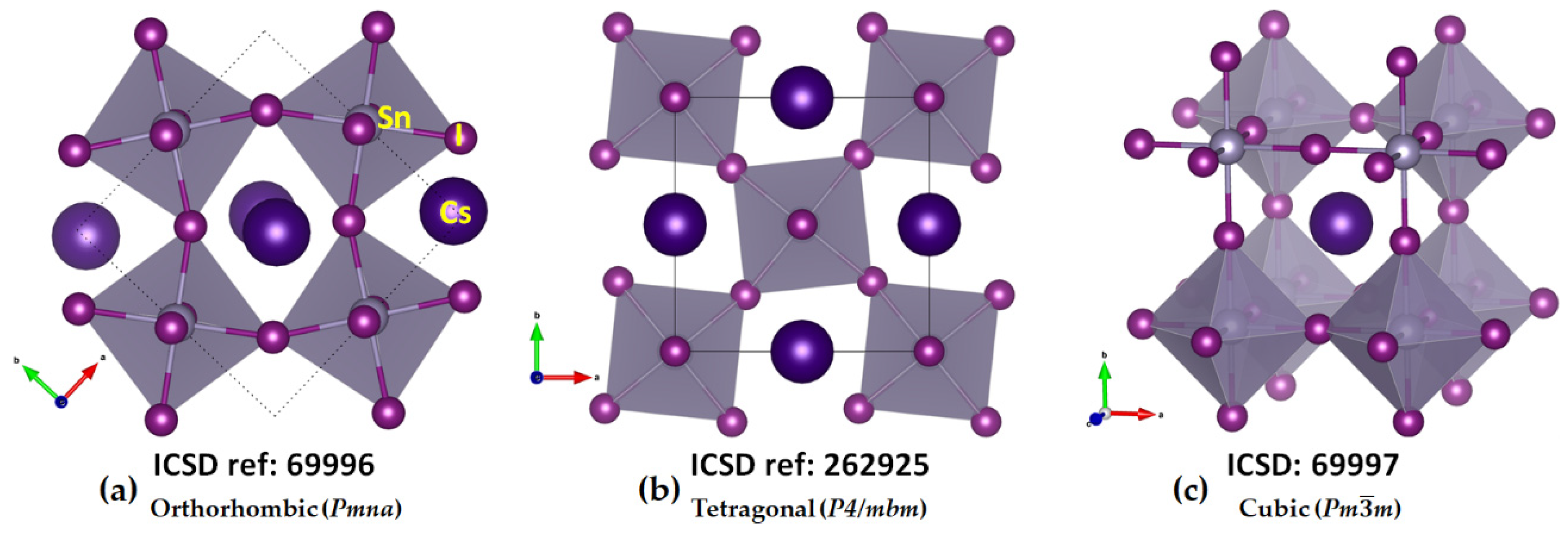{kind=link}
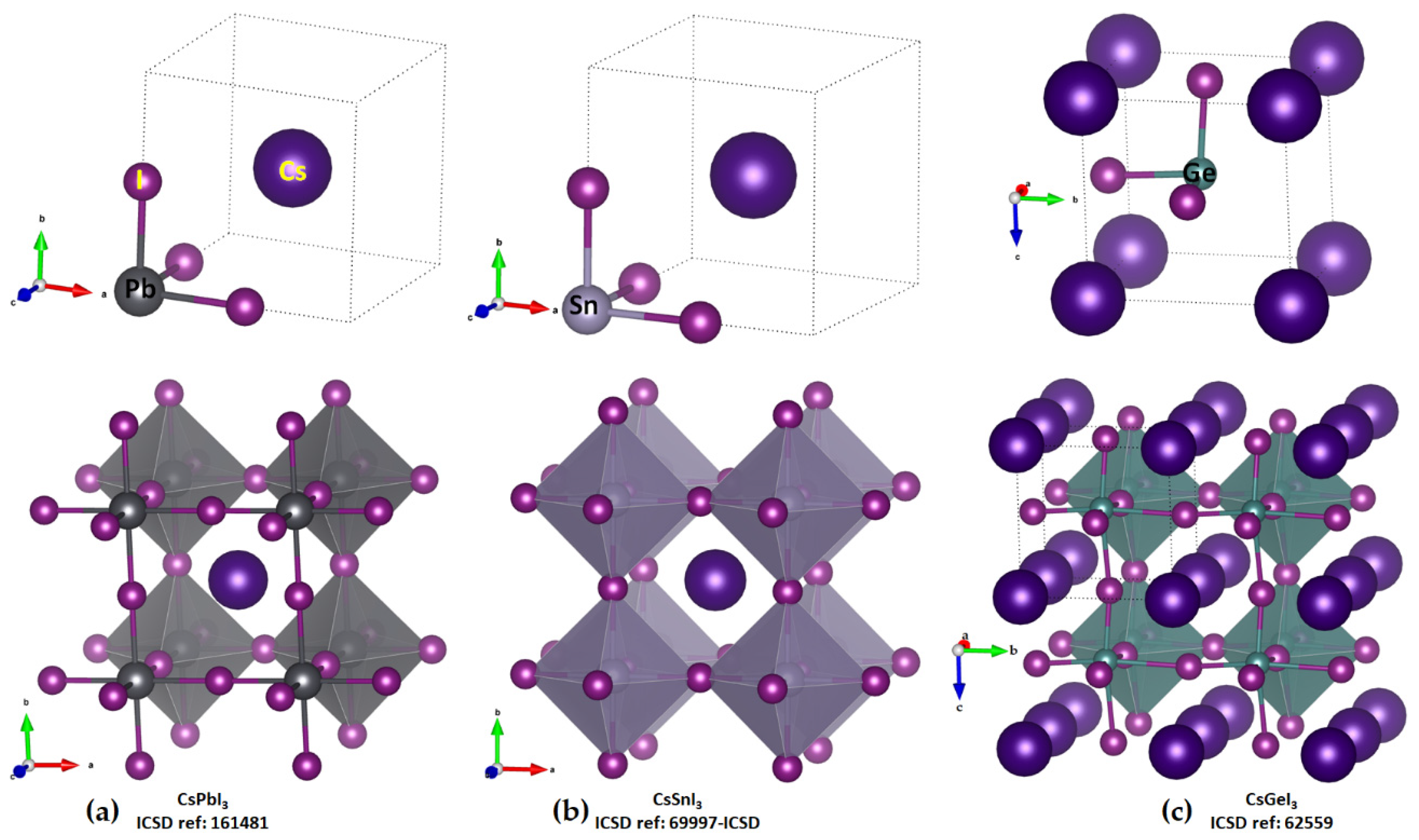{kind=link}
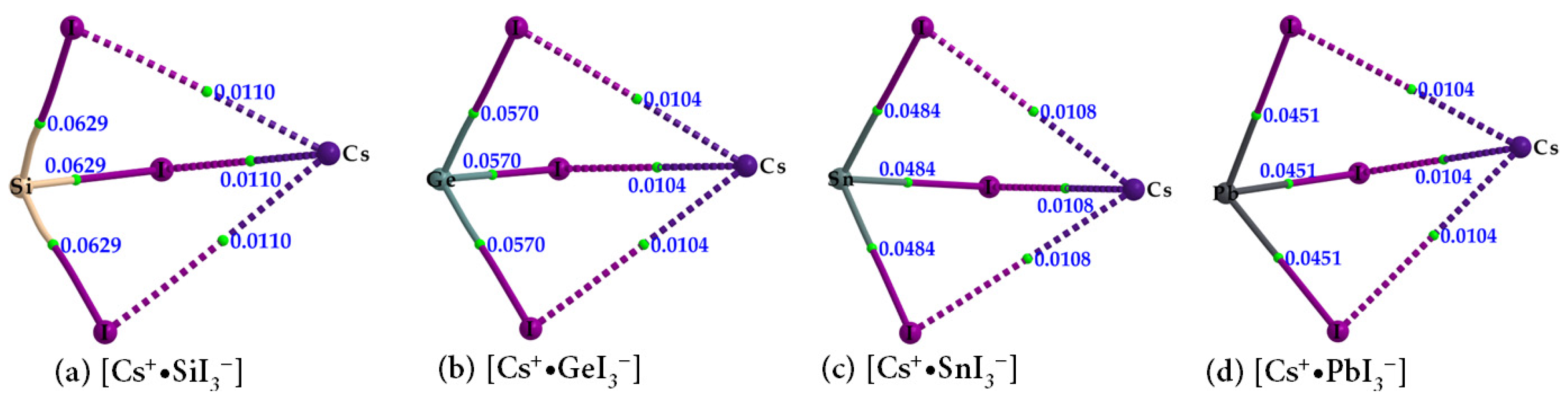{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}