
Expression Quantitative Trait Methylation Analysis Identifies Whole Blood Molecular Footprint in Fetal Alcohol Spectrum Disorder (FASD)
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Study Cohort
2.2. DNA Methylation Data
2.3. Epigenome-Wide Loci Associated with FASD
2.4. Absent Differential Gene Expression in Whole Blood-Derived RNA of Individuals with FASD
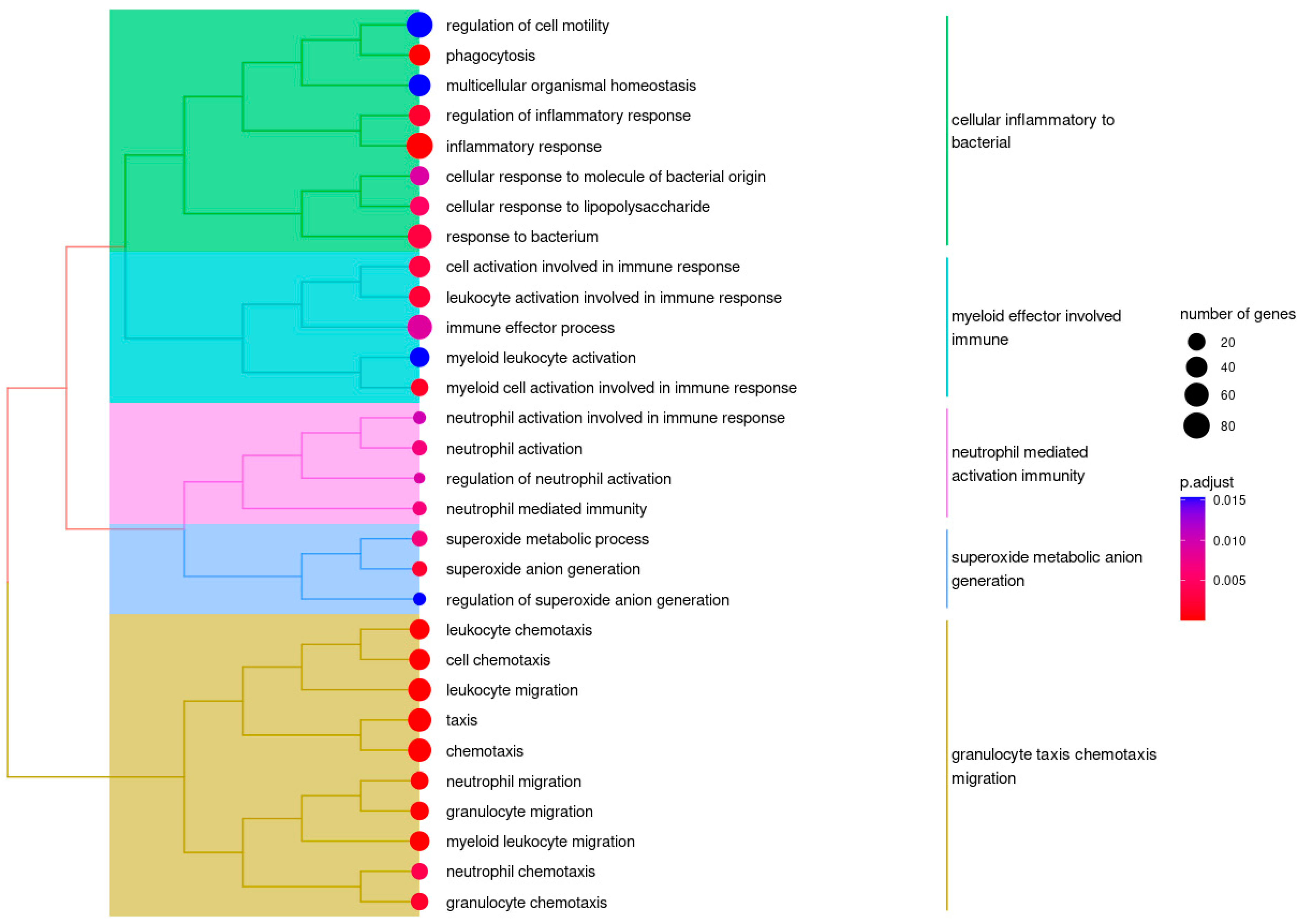
2.5. Weighted Correlation Network Analysis Identified Clusters of Genes Associated with FASD
2.6. Cis-Regulatory DNAm Elements Associated with FASD
3. Discussion
4. Materials and Methods
4.1. Subject and Sample Collection
4.2. Bioinformatics Analyses
4.3. DNA Methylation Profiling and Analysis
4.4. RNA Sequencing and Analysis
4.5. Weighted Correlation Network Analysis
4.6. Expression Quantitiative Trait Mehtylation (eQTM) Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Astley, S.J.; Clarren, S.K. Diagnosing the full spectrum of fetal alcohol-exposed individuals: Introducing the 4-digit diagnostic code. Alcohol Alcohol. 2000, 35, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, J.R.; Riley, E.P.; Charness, M.E. Clinical presentation, diagnosis, and management of fetal alcohol spectrum disorder. Lancet Neurol. 2019, 18, 760–770. [Google Scholar] [CrossRef]
- Chasnoff, I.J.; Wells, A.M.; King, L. Misdiagnosis and missed diagnoses in foster and adopted children with prenatal alcohol exposure. Pediatrics 2015, 135, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Cobben, J.M.; Krzyzewska, I.M.; Venema, A.; Mul, A.N.; Polstra, A.; Postma, A.V.; Smigiel, R.; Pesz, K.; Niklinski, J.; Chomczyk, M.A.; et al. DNA methylation abundantly associates with fetal alcohol spectrum disorder and its subphenotypes. Epigenomics 2019, 11, 767–785. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [Green Version]
- van Iterson, M.; Tobi, E.W.; Slieker, R.C.; den Hollander, W.; Luijk, R.; Slagboom, P.E.; Heijmans, B.T. MethylAid: Visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 2014, 30, 3435–3437. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, T.W. Prenatal Alcohol Exposure and the Developing Immune System. Alcohol. Res. 2015, 37, 279–285. [Google Scholar]
- Lussier, A.A.; Morin, A.M.; MacIsaac, J.L.; Salmon, J.; Weinberg, J.; Reynolds, J.N.; Pavlidis, P.; Chudley, A.E.; Kobor, M.S. DNA methylation as a predictor of fetal alcohol spectrum disorder. Clin. Epigenetics 2018, 10, 5. [Google Scholar] [CrossRef]
- Portales-Casamar, E.; Lussier, A.A.; Jones, M.J.; MacIsaac, J.L.; Edgar, R.D.; Mah, S.M.; Barhdadi, A.; Provost, S.; Lemieux-Perreault, L.P.; Cynader, M.S.; et al. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin 2016, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Wilson, L.E.; Xu, Z.; Harlid, S.; White, A.J.; Troester, M.A.; Sandler, D.P.; Taylor, J.A. Alcohol and DNA Methylation: An Epigenome-Wide Association Study in Blood and Normal Breast Tissue. Am. J. Epidemiol. 2019, 188, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Gillentine, M.A.; Wang, T.; Hoekzema, K.; Rosenfeld, J.; Liu, P.; Guo, H.; Kim, C.N.; De Vries, B.B.A.; Vissers, L.; Nordenskjold, M.; et al. Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med. 2021, 13, 63. [Google Scholar] [CrossRef]
- Nickels, E.M.; Li, S.; Morimoto, L.; Kang, A.Y.; de Smith, A.J.; Metayer, C.; Wiemels, J.L. Periconceptional folate intake influences DNA methylation at birth based on dietary source in an analysis of pediatric acute lymphoblastic leukemia cases and controls. Am. J. Clin. Nutr. 2022, 116, 1553–1564. [Google Scholar] [CrossRef]
- Young, J.K.; Giesbrecht, H.E.; Eskin, M.N.; Aliani, M.; Suh, M. Nutrition implications for fetal alcohol spectrum disorder. Adv. Nutr. 2014, 5, 675–692. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, G.; Borrás-Novell, C.; Casanova, M.A.; Pascual Tutusaus, M.; Ferrero Martínez, S.; Gómez Roig, M.D.; García-Algar, O. The Effects of Alcohol and Drugs of Abuse on Maternal Nutritional Profile during Pregnancy. Nutrients 2018, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Harris, B.S.; Bishop, K.C.; Kemeny, H.R.; Walker, J.S.; Rhee, E.; Kuller, J.A. Risk Factors for Birth Defects. Obs. Gynecol. Surv. 2017, 72, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Ballard, M.S.; Sun, M.; Ko, J. Vitamin A, folate, and choline as a possible preventive intervention to fetal alcohol syndrome. Med. Hypotheses 2012, 78, 489–493. [Google Scholar] [CrossRef]
- Gillis, R.F.; Palmour, R.M. mRNA expression analysis of the hippocampus in a vervet monkey model of fetal alcohol spectrum disorder. J. Neurodev. Disord. 2022, 14, 21. [Google Scholar] [CrossRef]
- Lussier, A.A.; Bodnar, T.S.; Weinberg, J. Intersection of Epigenetic and Immune Alterations: Implications for Fetal Alcohol Spectrum Disorder and Mental Health. Front Neurosci. 2021, 15, 788630. [Google Scholar] [CrossRef]
- Gunasekara, C.J.; Scott, C.A.; Laritsky, E.; Baker, M.S.; MacKay, H.; Duryea, J.D.; Kessler, N.J.; Hellenthal, G.; Wood, A.C.; Hodges, K.R.; et al. A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biol. 2019, 20, 105. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Arzua, T.; Yan, Y.; Jiang, C.; Logan, S.; Allison, R.L.; Wells, C.; Kumar, S.N.; Schäfer, R.; Bai, X. Modeling alcohol-induced neurotoxicity using human induced pluripotent stem cell-derived three-dimensional cerebral organoids. Transl. Psychiatry 2020, 10, 347. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, L.; Yin, F.; Yu, Y.; Wang, Y.; Shepard, M.J.; Zhuang, Z.; Qin, J. Probing impaired neurogenesis in human brain organoids exposed to alcohol. Integr. Biol. (Camb) 2017, 9, 968–978. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Reginald, V.L.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.; Fast, Q.C. A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 July 2022).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Li Yim, A.Y.F.; de Bruyn, J.R.; Duijvis, N.W.; Sharp, C.; Ferrero, E.; de Jonge, W.J.; Wildenberg, M.E.; Mannens, M.; Buskens, C.J.; D’Haens, G.R.; et al. A distinct epigenetic profile distinguishes stenotic from non-inflamed fibroblasts in the ileal mucosa of Crohn’s disease patients. PLoS ONE 2018, 13, e0209656. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
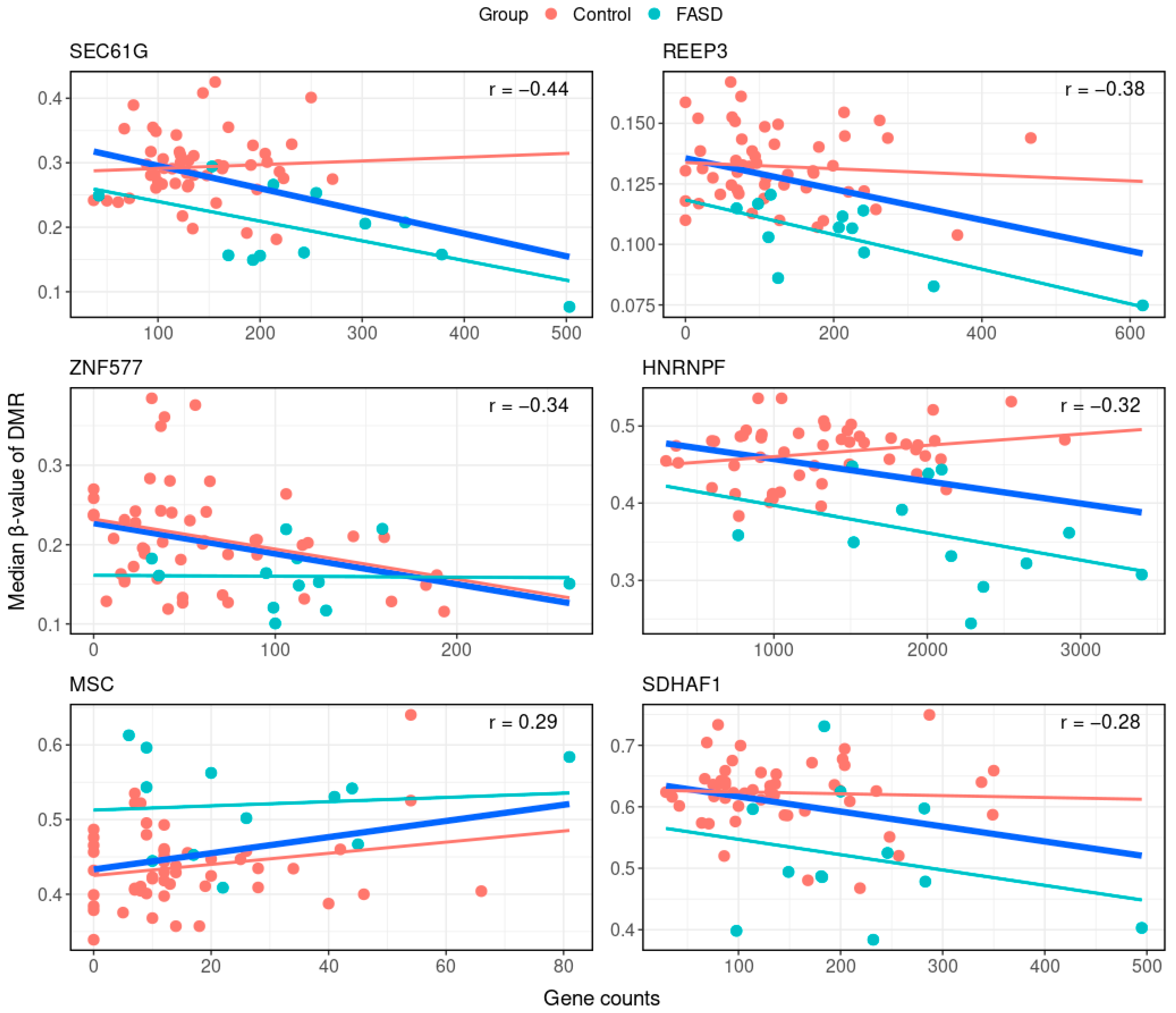
| Position | Gene | Correlation Coefficient | p Value | DMR Feature | DMR Direction | Gene Expression Direction |
|---|---|---|---|---|---|---|
| chr7:54827528-54827677 | SEC61G | −0.44 | 2.29 × 10−3 | Promoter | Hypo-methylated | Overexpression |
| chr10:65280473-65280961 | REEP3 | −0.38 | 3.69 × 10−3 | Promoter | Hypo-methylated | Overexpression |
| chr19:52391078-52391090 | ZNF577 | −0.34 | 8.33 × 10−3 | 1st exon | Hypo-methylated | Overexpression |
| chr10:43891459-43892075 | HNRNPF | −0.32 | 1.55 × 10−2 | Gene body | Hypo-methylated | Overexpression |
| chr8:72758461-72758701 | MSC | 0.29 | 1.98 × 10−2 | Promoter | Hyper-methylated | Overexpression |
| chr19:36484731-36485360 | SDHAF1 | −0.28 | 3.12 × 10−2 | Promoter | Hypo-methylated | Overexpression |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krzyzewska, I.M.; Lauffer, P.; Mul, A.N.; van der Laan, L.; Yim, A.Y.F.L.; Cobben, J.M.; Niklinski, J.; Chomczyk, M.A.; Smigiel, R.; Mannens, M.M.A.M.; et al. Expression Quantitative Trait Methylation Analysis Identifies Whole Blood Molecular Footprint in Fetal Alcohol Spectrum Disorder (FASD). Int. J. Mol. Sci. 2023, 24, 6601. https://doi.org/10.3390/ijms24076601
Krzyzewska IM, Lauffer P, Mul AN, van der Laan L, Yim AYFL, Cobben JM, Niklinski J, Chomczyk MA, Smigiel R, Mannens MMAM, et al. Expression Quantitative Trait Methylation Analysis Identifies Whole Blood Molecular Footprint in Fetal Alcohol Spectrum Disorder (FASD). International Journal of Molecular Sciences. 2023; 24(7):6601. https://doi.org/10.3390/ijms24076601
Chicago/Turabian StyleKrzyzewska, Izabela M., Peter Lauffer, Adri N. Mul, Liselot van der Laan, Andrew Y. F. Li Yim, Jan Maarten Cobben, Jacek Niklinski, Monika A. Chomczyk, Robert Smigiel, Marcel M. A. M. Mannens, and et al. 2023. "Expression Quantitative Trait Methylation Analysis Identifies Whole Blood Molecular Footprint in Fetal Alcohol Spectrum Disorder (FASD)" International Journal of Molecular Sciences 24, no. 7: 6601. https://doi.org/10.3390/ijms24076601