
Myokine Musclin Is Critical for Exercise-Induced Cardiac Conditioning
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
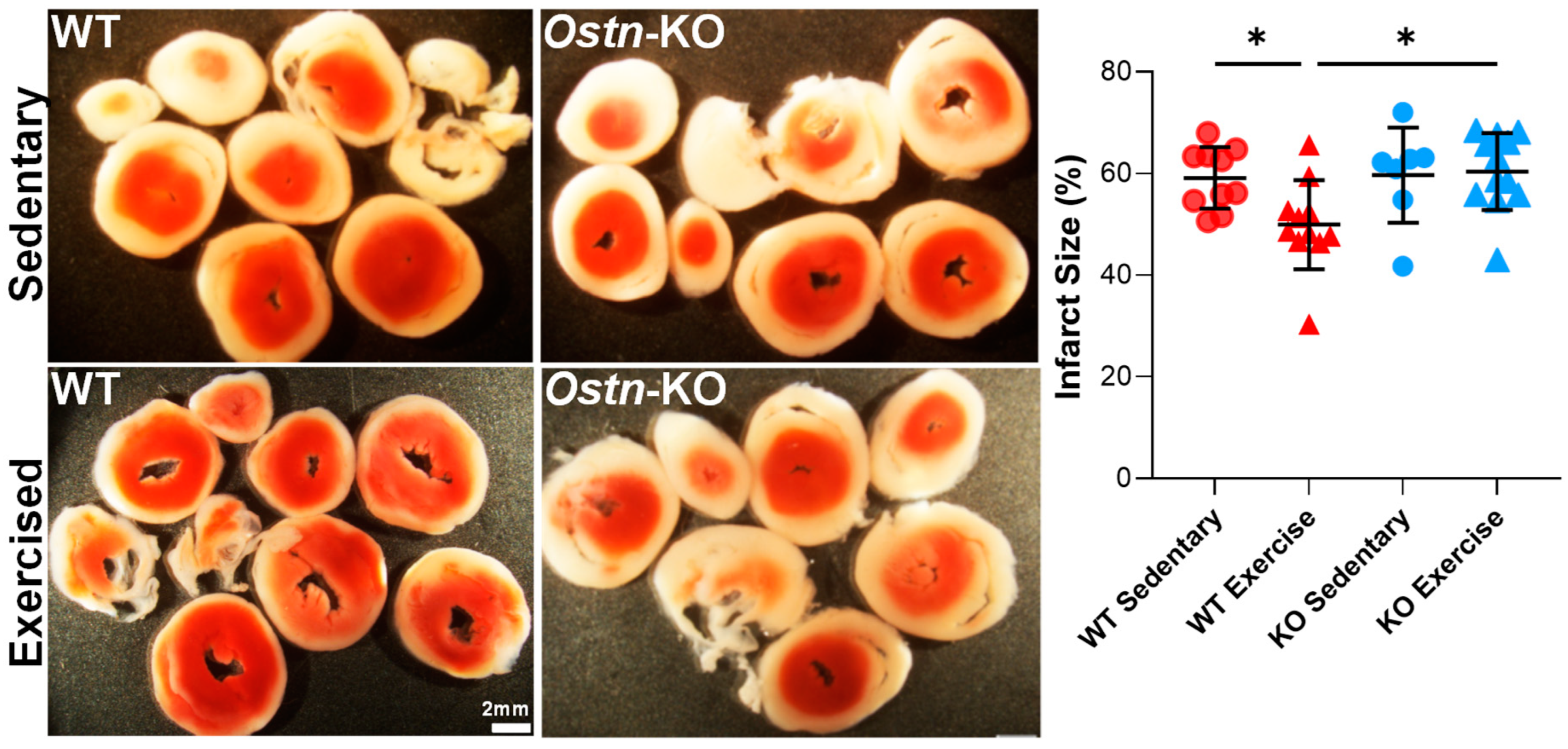
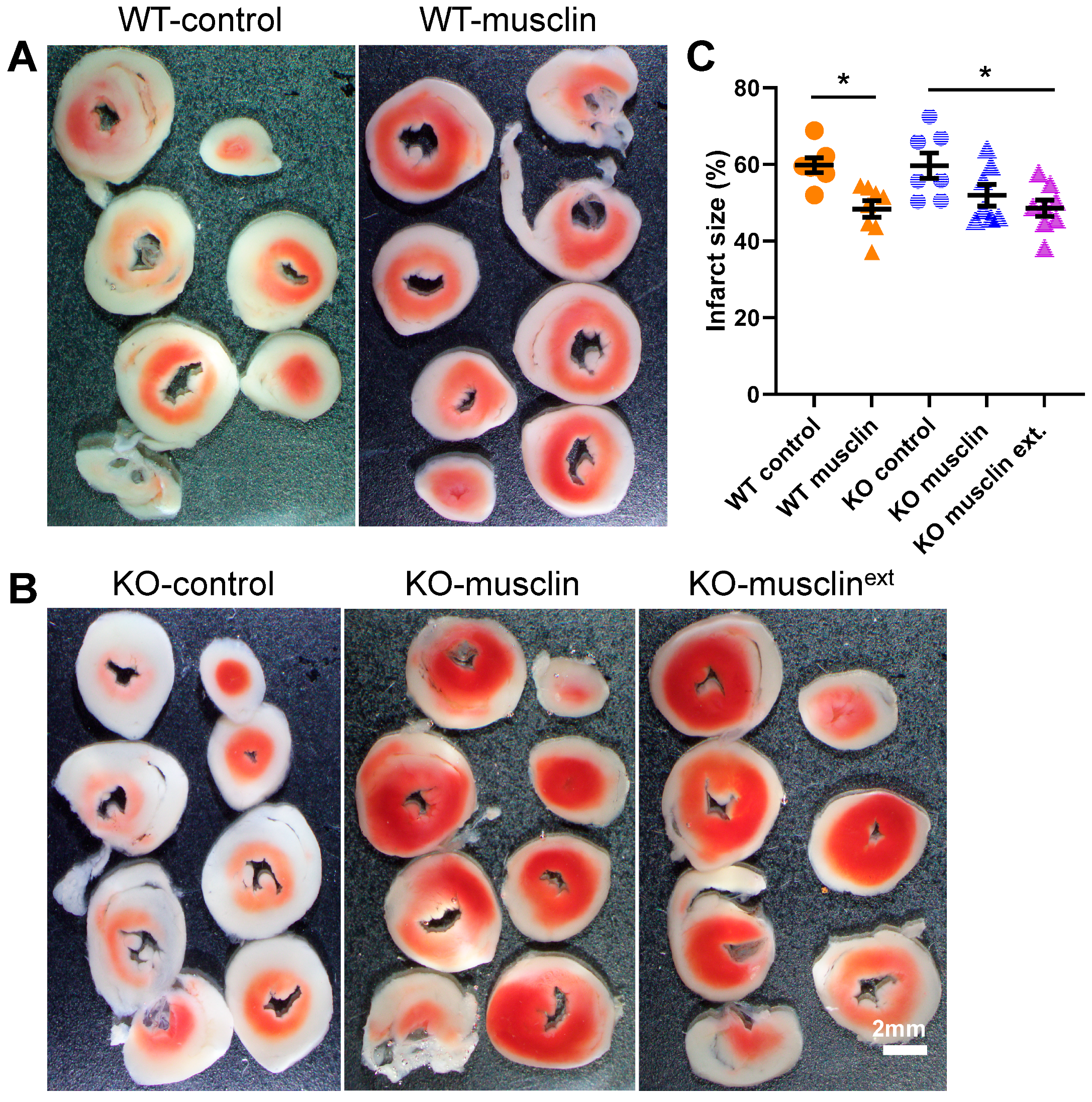
2.1. Musclin Is Required for Exercise-Mediated Protection from Ischemia-Reperfusion Injury
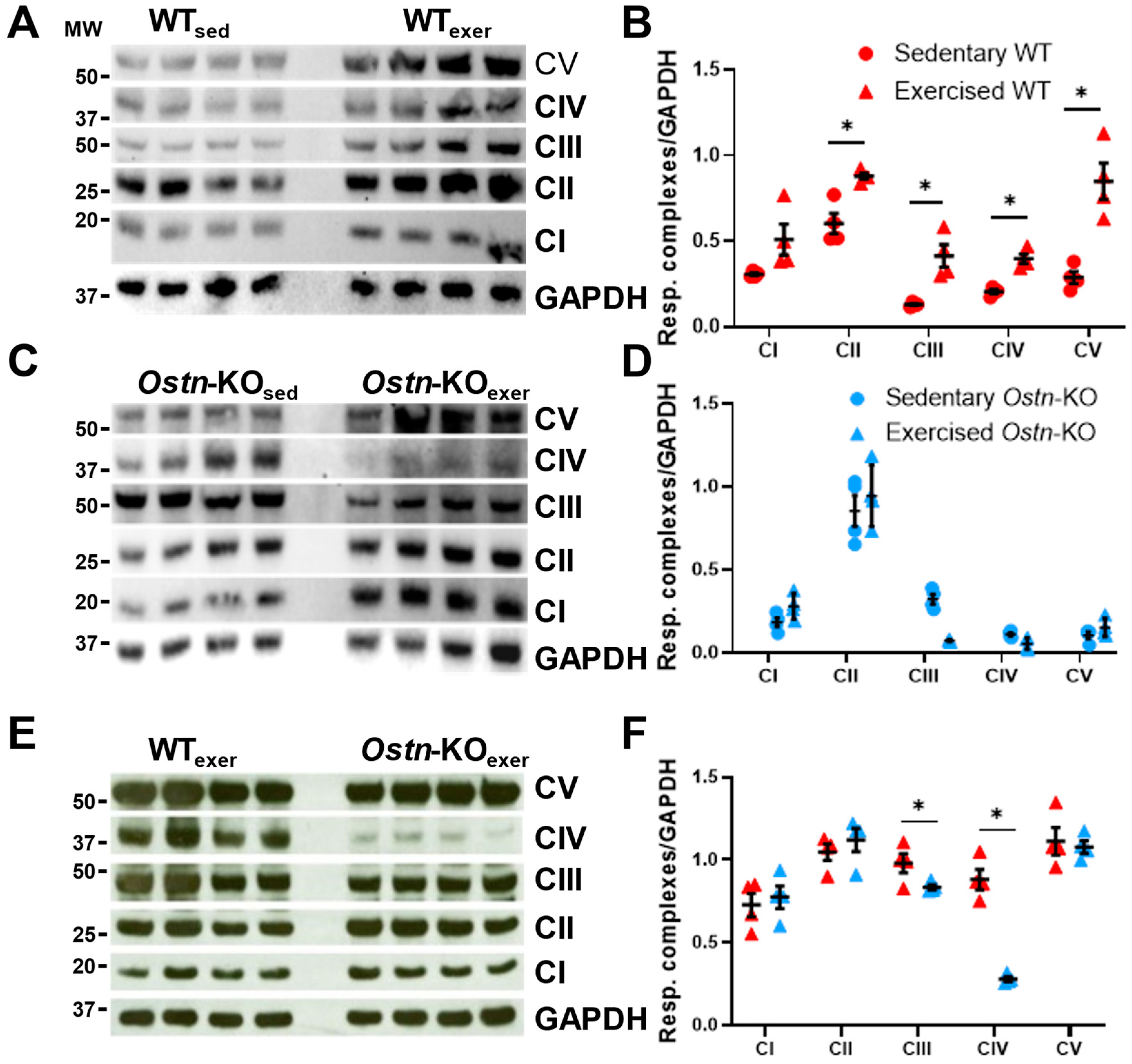
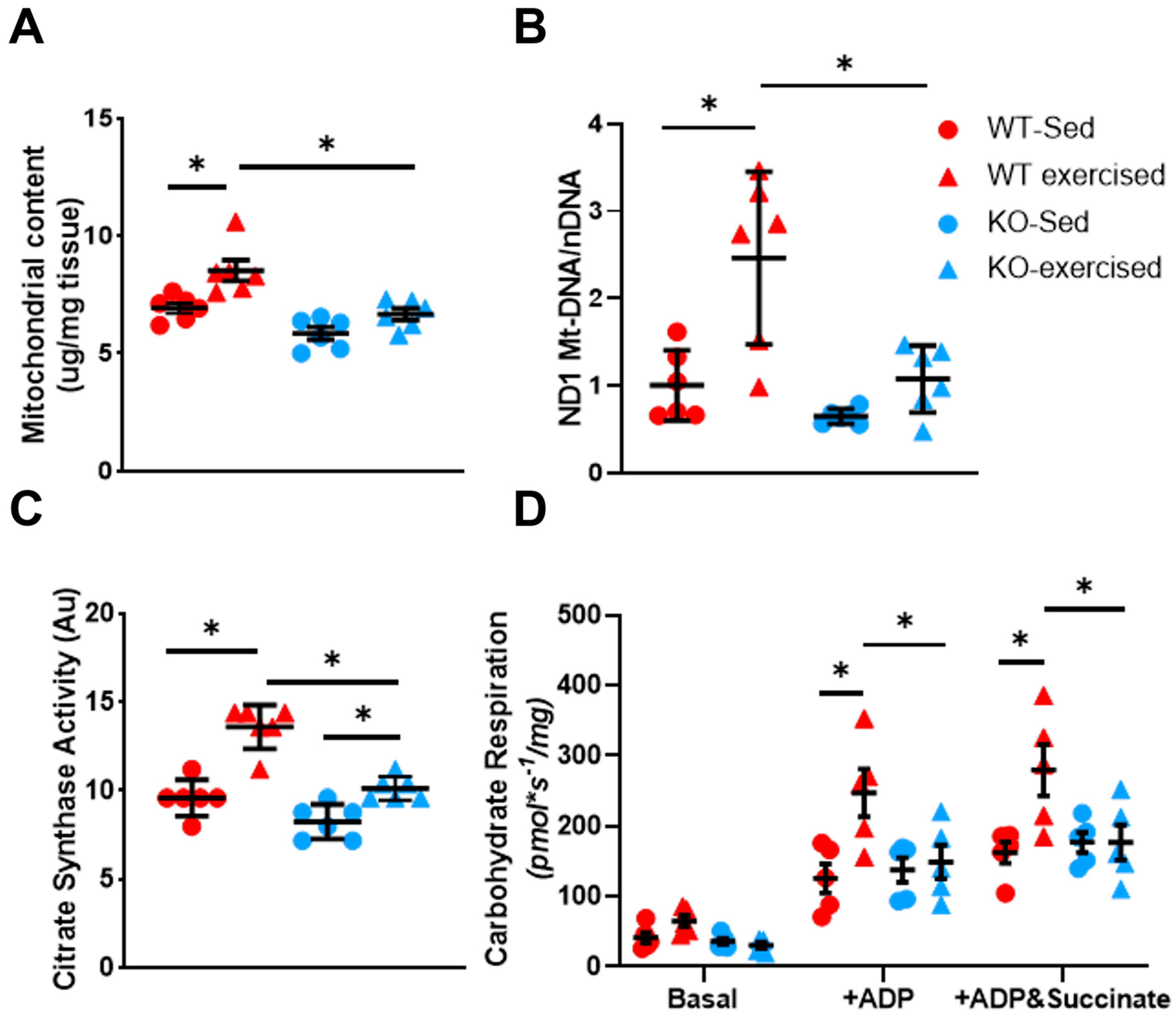
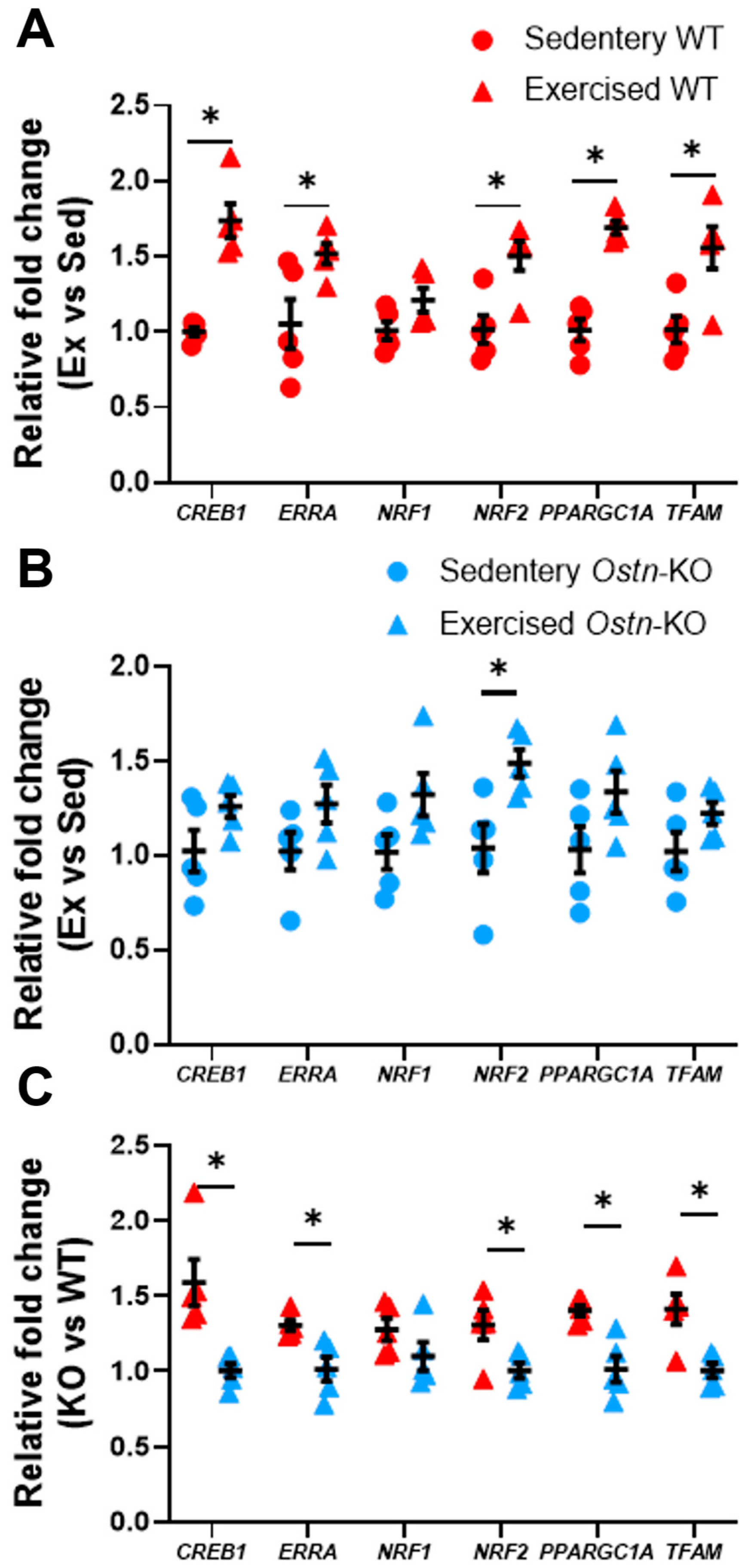
2.2. Musclin Promotes Exercise Training-Induced Enhancements in Mitochondrial Biogenesis and Respiratory Capacity
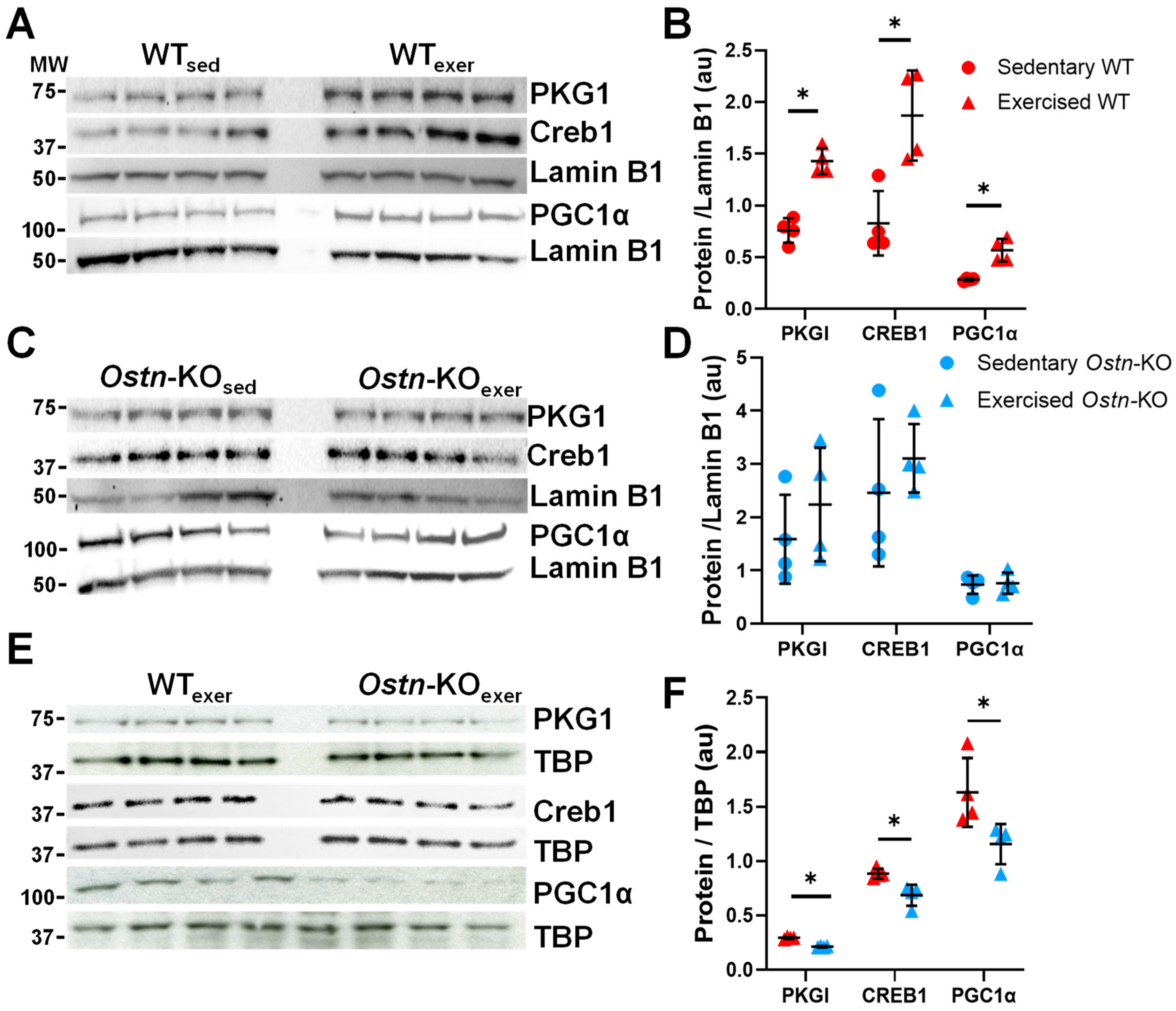
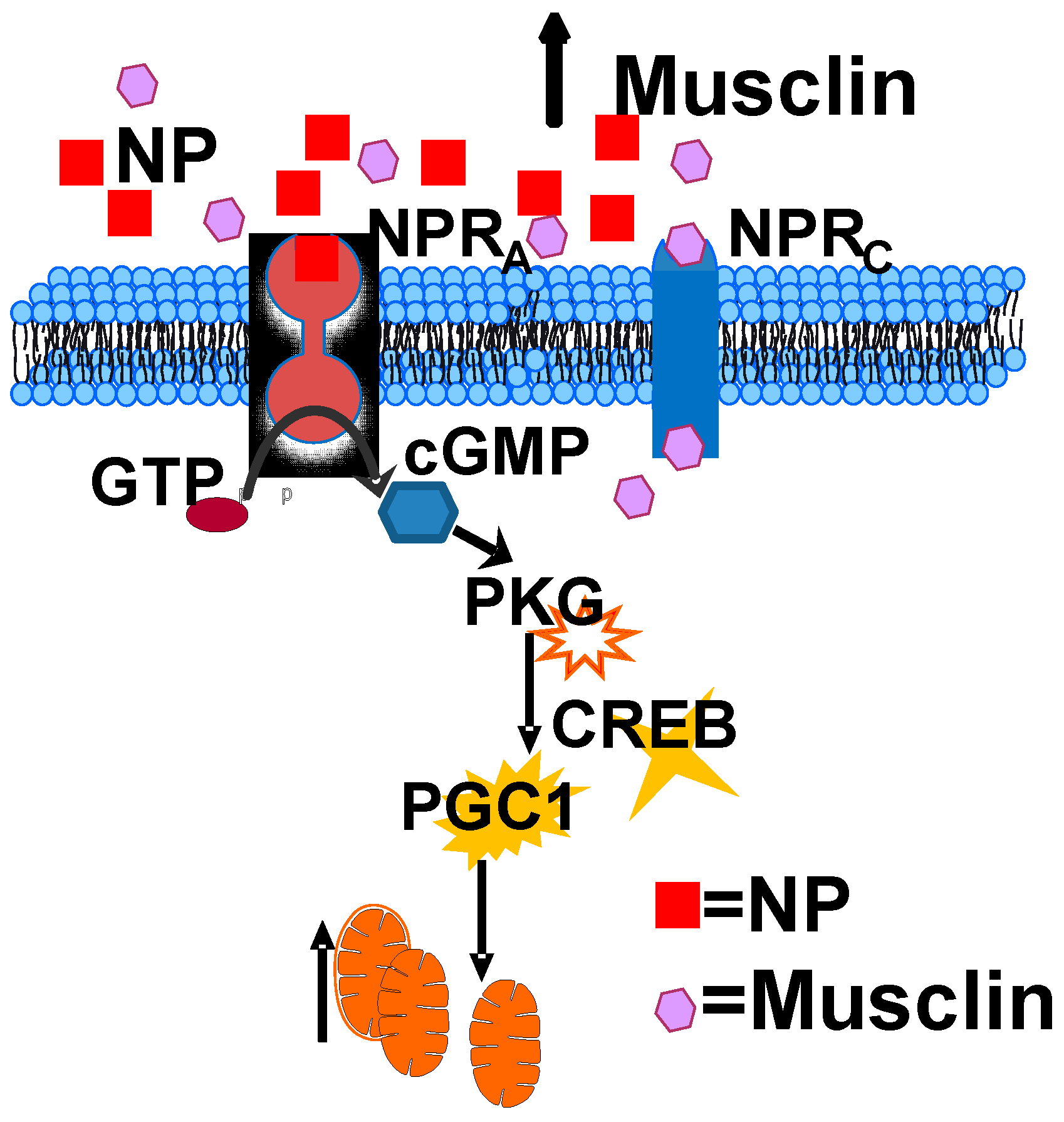
2.3. Musclin Regulates Cardiac cGMP/Protein Kinase G-Dependent Signaling
2.4. Synthetic Musclin Infusion Mimics Exercise-Mediated Protection against IR Injury
3. Discussion
4. Methods
4.1. Animal Experiments
4.2. Ostn-KO Mouse Model
4.3. Myocardial Ischemia–Reperfusion Injury
4.4. Treadmill Exercise Training
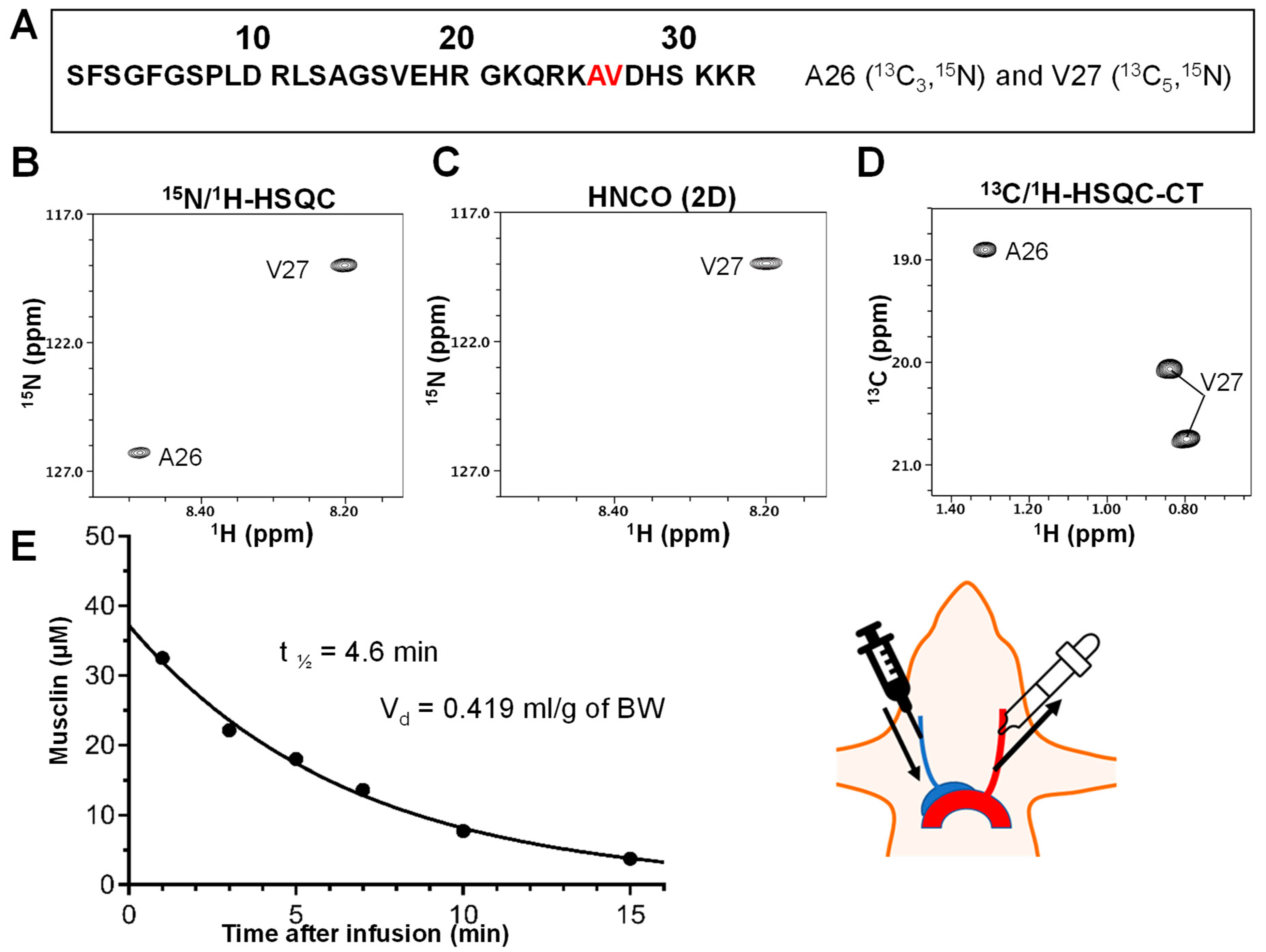
4.5. Determination of Synthetic Musclin Peptide Plasma Elimination Half-Life and Volume of Distribution
4.6. NMR Spectroscopy
4.7. Pharmacokinetic Calculations
- Peptide MW: 3649.675
- Initial concentration: 36 µM = 0.131 mg/mL
- Q—infusion rate
- Css—steady state concentration
- Cl—clearance
- T1/2—half-life
- Ke—elimination rate = 0.693/T1/2 = 0.151 min−1
- Vd—volume of distribution = administered dose/initial concentration
- 1.7 mg/0.131 mg/mL = 12.997 mL for mice of 31 g → 0.419 mL/g of BW
- Desired steady state concentration: 200 pg/mL
- Cl = Ke × Vd = 0.151 min−1 × 0.419 mL/g × 27 g = 1.71 mL/min
- Q = Css × Cl = 200 pg/mL × 1.71 mL/min = 341.652 pg/min → 20.28 ng/h
- For mice of 27 g:
- Q = 174.636 pg/min → 10.478 ng/h → 3520.608 ng for 2 weeks
- For osmotic pumps with 0.25 µL/h → 20.28 × 4 = 81.966 ng/µL → 8.197 µg per pump
4.8. Osmotic Pumps
4.9. Mitochondrial Protein Isolation
4.10. RNA Isolation and qRT-PCR
4.11. Western Blotting
4.12. CGMP Measurement
4.13. Mitochondrial Respiration in Isolated Myofibers
4.14. Mitochondrial DNA
4.15. Citrate Synthase Activity
4.16. Statistics
5. Conclusions
- The exercise-responsive myokine musclin, which has high homology to natriuretic peptides (NPs), is critical for exercise-induced myocardial protection.
- Normal musclin signaling is critical for exercise-induced cardiac mitochondrial biogenesis.
- Musclin-dependent cardioprotective remodeling is driven by activation of cGMP/PKGI/CREB/PGC1α signaling.
- Uncovering a musclin-dependent effect on exercise-induced cardiac conditioning broadens the appreciation of the complexity and physiologic roles of cardiac and skeletal muscles as endocrine organs.
- Synthetic musclin infusion reproduces the exercise-induced cardioprotective response to ischemia-reperfusion.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| ANOVA | analysis of variance |
| ANP | atrial natriuretic peptide |
| BNP | brain natriuretic peptide |
| cGMP | cyclic guanosine monophosphate |
| CREB | cyclic adenosine monophosphate response element binding protein |
| CS | citrate synthase |
| DNA | deoxyribonucleic acid |
| ERRA | estrogen related receptor alpha |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| HPRT | hypoxanthine-guanine phosphoribosyltransferase |
| IPC | ischemic pre-conditioning |
| IR | ischemia/reperfusion |
| MI | myocardial infarction |
| ND1 | NADH dehydrogenase 1 |
| NMR | nuclear magnetic resonance |
| NP | natriuretic peptide |
| NPR | natriuretic peptide receptor |
| NRF1 | nuclear respiratory factor 1 |
| NRF2 | nuclear respiratory factor 2 |
| OSTN | osteocrin |
| OXPHOS | oxidative phosphorylation |
| PGC1α | peroxisome proliferator-activated receptor γ coactivator 1 alpha |
| PKG | cyclic GMP-dependent protein kinase |
| PPAR | peroxisome proliferator activated receptor |
| RNA | ribonucleic acid |
| SD | standard deviation |
| TBP | TATA-binding protein |
| TFAM | transcription factor a, mitochondrial |
| TTC | triphenyltetrazolium chloride |
References
- Masaebi, F.; Salehi, M.; Kazemi, M.; Vahabi, N.; Azizmohammad Looha, M.; Zayeri, F. Trend analysis of disability adjusted life years due to cardiovascular diseases: Results from the global burden of disease study 2019. BMC Public Health 2021, 21, 1268. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update from the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- McMurray, J.J. Clinical practice. Systolic heart failure. N. Engl. J. Med. 2010, 362, 228–238. [Google Scholar] [CrossRef]
- Cokkinos, D.V.; Pantos, C. Myocardial protection in man—From research concept to clinical practice. Heart Fail. Rev. 2007, 12, 345–362. [Google Scholar] [CrossRef]
- Juhaszova, M.; Rabuel, C.; Zorov, D.B.; Lakatta, E.G.; Sollott, S.J. Protection in the aged heart: Preventing the heart-break of old age? Cardiovasc. Res. 2005, 66, 233–244. [Google Scholar] [CrossRef]
- Eijsvogels, T.M.; Molossi, S.; Lee, D.C.; Emery, M.S.; Thompson, P.D. Exercise at the Extremes: The Amount of Exercise to Reduce Cardiovascular Events. J. Am. Coll. Cardiol. 2016, 67, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Pina, I.L.; Apstein, C.S.; Balady, G.J.; Belardinelli, R.; Chaitman, B.R.; Duscha, B.D.; Fletcher, B.J.; Fleg, J.L.; Myers, J.N.; Sullivan, M.J.; et al. Exercise and heart failure: A statement from the American Heart Association Committee on exercise, rehabilitation, and prevention. Circulation 2003, 107, 1210–1225. [Google Scholar] [CrossRef]
- Thompson, P.D.; Buchner, D.; Pina, I.L.; Balady, G.J.; Williams, M.A.; Marcus, B.H.; Berra, K.; Blair, S.N.; Costa, F.; Franklin, B.; et al. Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease: A statement from the Council on Clinical Cardiology (Subcommittee on Exercise, Rehabilitation, and Prevention) and the Council on Nutrition, Physical Activity, and Metabolism (Subcommittee on Physical Activity). Circulation 2003, 107, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Vujic, A.; Lerchenmuller, C.; Wu, T.D.; Guillermier, C.; Rabolli, C.P.; Gonzalez, E.; Senyo, S.E.; Liu, X.; Guerquin-Kern, J.L.; Steinhauser, M.L.; et al. Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat. Commun. 2018, 9, 1659. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.; Oldridge, N.; Thompson, D.R.; Zwisler, A.D.; Rees, K.; Martin, N.; Taylor, R.S. Exercise-Based Cardiac Rehabilitation for Coronary Heart Disease: Cochrane Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2016, 67, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puetz, T.W.; Beasman, K.M.; O’Connor, P.J. The effect of cardiac rehabilitation exercise programs on feelings of energy and fatigue: A meta-analysis of research from 1945 to 2005. Eur. J. Cardiovasc. Prev. Rehabil. 2006, 13, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason-Wehrens, B.; Nebel, R.; Jensen, K.; Hackbusch, M.; Grilli, M.; Gielen, S.; Schwaab, B.; Rauch, B. Exercise-based cardiac rehabilitation in patients with reduced left ventricular ejection fraction: The Cardiac Rehabilitation Outcome Study in Heart Failure (CROS-HF): A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2019, 27, 2047487319854140. [Google Scholar] [CrossRef] [PubMed]
- Mezzani, A.; Hamm, L.F.; Jones, A.M.; McBride, P.E.; Moholdt, T.; Stone, J.A.; Urhausen, A.; Williams, M.A. Aerobic exercise intensity assessment and prescription in cardiac rehabilitation: A joint position statement of the European Association for Cardiovascular Prevention and Rehabilitation, the American Association of Cardiovascular and Pulmonary Rehabilitation and the Canadian Association of Cardiac Rehabilitation. Eur. J. Prev. Cardiol. 2013, 20, 442–467. [Google Scholar] [CrossRef]
- Powers, S.K. Exercise: Teaching myocytes new tricks. J. Appl. Physiol. 2017, 123, 460–472. [Google Scholar] [CrossRef] [Green Version]
- Hulzebos, E.H.; Smit, Y.; Helders, P.P.; van Meeteren, N.L. Preoperative physical therapy for elective cardiac surgery patients. Cochrane Database Syst. Rev. 2012, 11, CD010118. [Google Scholar] [CrossRef]
- Lemanu, D.P.; Singh, P.P.; MacCormick, A.D.; Arroll, B.; Hill, A.G. Effect of preoperative exercise on cardiorespiratory function and recovery after surgery: A systematic review. World J. Surg. 2013, 37, 711–720. [Google Scholar] [CrossRef]
- Hulzebos, E.H.; Helders, P.J.; Favie, N.J.; De Bie, R.A.; Brutel de la Riviere, A.; Van Meeteren, N.L. Preoperative intensive inspiratory muscle training to prevent postoperative pulmonary complications in high-risk patients undergoing CABG surgery: A randomized clinical trial. JAMA 2006, 296, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Hoogeboom, T.J.; Dronkers, J.J.; Hulzebos, E.H.; van Meeteren, N.L. Merits of exercise therapy before and after major surgery. Curr. Opin. Anaesthesiol. 2014, 27, 161–166. [Google Scholar] [CrossRef]
- Zhu, Z.; Gao, Z.; Chen, B.; Hall, D.D.; Minerath, R.; Koval, O.; Sierra, A.; Subbotina, E.; Zhu, X.; Kim, Y.R.; et al. Atrial-paced, exercise-similar heart rate envelope induces myocardial protection from ischaemic injury. EP Eur. 2021, 24, 1025–1035. [Google Scholar] [CrossRef]
- Chow, L.S.; Gerszten, R.E.; Taylor, J.M.; Pedersen, B.K.; van Praag, H.; Trappe, S.; Febbraio, M.A.; Galis, Z.S.; Gao, Y.; Haus, J.M.; et al. Exerkines in health, resilience and disease. Nat. Rev. Endocrinol. 2022, 18, 273–289. [Google Scholar] [CrossRef]
- Sabaratnam, R.; Wojtaszewski, J.F.P.; Hojlund, K. Factors mediating exercise-induced organ crosstalk. Acta Physiol. 2022, 234, e13766. [Google Scholar] [CrossRef] [PubMed]
- Subbotina, E.; Sierra, A.; Zhu, Z.; Gao, Z.; Koganti, S.R.; Reyes, S.; Stepniak, E.; Walsh, S.A.; Acevedo, M.R.; Perez-Terzic, C.M.; et al. Musclin is an activity-stimulated myokine that enhances physical endurance. Proc. Natl. Acad. Sci. USA 2015, 112, 16042–16047. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Otani, K.; Chiba, A.; Nishimura, H.; Tokudome, T.; Takano-Watanabe, H.; Matsuo, A.; Ishikawa, H.; Shimamoto, K.; Fukui, H.; et al. A New Secretory Peptide of Natriuretic Peptide Family, Osteocrin, Suppresses the Progression of Congestive Heart Failure After Myocardial Infarction. Circ. Res. 2018, 122, 742–751. [Google Scholar] [CrossRef]
- Szaroszyk, M.; Kattih, B.; Martin-Garrido, A.; Trogisch, F.A.; Dittrich, G.M.; Grund, A.; Abouissa, A.; Derlin, K.; Meier, M.; Holler, T.; et al. Skeletal muscle derived Musclin protects the heart during pathological overload. Nat. Commun. 2022, 13, 149. [Google Scholar] [CrossRef]
- Pollen, A.A.; Kriegstein, A.R. Primate Neurons Flex Their Musclin. Neuron 2016, 92, 681–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ataman, B.; Boulting, G.L.; Harmin, D.A.; Yang, M.G.; Baker-Salisbury, M.; Yap, E.L.; Malik, A.N.; Mei, K.; Rubin, A.A.; Spiegel, I.; et al. Evolution of Osteocrin as an activity-regulated factor in the primate brain. Nature 2016, 539, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, A.; Watanabe-Takano, H.; Terai, K.; Fukui, H.; Miyazaki, T.; Uemura, M.; Hashimoto, H.; Hibi, M.; Fukuhara, S.; Mochizuki, N. Osteocrin, a peptide secreted from the heart and other tissues, contributes to cranial osteogenesis and chondrogenesis in zebrafish. Development 2017, 144, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.; Moffatt, P.; Salois, P.; Gaumond, M.H.; Gingras, R.; Godin, E.; Miao, D.; Goltzman, D.; Lanctot, C. Osteocrin, a novel bone-specific secreted protein that modulates the osteoblast phenotype. J. Biol. Chem. 2003, 278, 50563–50571. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, H.; Matsuda, M.; Yamada, Y.; Kawai, K.; Suzuki, E.; Makishima, M.; Kitamura, T.; Shimomura, I. Musclin, a novel skeletal muscle-derived secretory factor. J. Biol. Chem. 2004, 279, 19391–19395. [Google Scholar] [CrossRef] [Green Version]
- Kita, S.; Nishizawa, H.; Okuno, Y.; Tanaka, M.; Yasui, A.; Matsuda, M.; Yamada, Y.; Shimomura, I. Competitive binding of musclin to natriuretic peptide receptor 3 with atrial natriuretic peptide. J. Endocrinol. 2009, 201, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, P.; Thomas, G.; Sellin, K.; Bessette, M.C.; Lafreniere, F.; Akhouayri, O.; St-Arnaud, R.; Lanctot, C. Osteocrin is a specific ligand of the natriuretic Peptide clearance receptor that modulates bone growth. J. Biol. Chem. 2007, 282, 36454–36462. [Google Scholar] [CrossRef] [Green Version]
- Bowles, D.K.; Starnes, J.W. Exercise training improves metabolic response after ischemia in isolated working rat heart. J. Appl. Physiol. 1994, 76, 1608–1614. [Google Scholar] [CrossRef]
- Demirel, H.A.; Powers, S.K.; Caillaud, C.; Coombes, J.S.; Naito, H.; Fletcher, L.A.; Vrabas, I.; Jessup, J.V.; Ji, L.L. Exercise training reduces myocardial lipid peroxidation following short-term ischemia-reperfusion. Med. Sci. Sport. Exerc. 1998, 30, 1211–1216. [Google Scholar] [CrossRef]
- Lennon, S.L.; Quindry, J.C.; French, J.P.; Kim, S.; Mehta, J.L.; Powers, S.K. Exercise and myocardial tolerance to ischaemia-reperfusion. Acta Physiol. Scand. 2004, 182, 161–169. [Google Scholar] [CrossRef]
- Theodorou, N.A.; Howell, S.L. An assessment of diffusion chambers for use in pancreatic islet cell transplantation. Transplantation 1979, 27, 350–352. [Google Scholar]
- Powers, S.K.; Lennon, S.L.; Quindry, J.; Mehta, J.L. Exercise and cardioprotection. Curr. Opin. Cardiol. 2002, 17, 495–502. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Zhang, X.Q.; Ng, Y.C.; Rothblum, L.I.; Musch, T.I.; Moore, R.L.; Cheung, J.Y. Sprint training normalizes Ca(2+) transients and SR function in postinfarction rat myocytes. J. Appl. Physiol. 2000, 89, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Quindry, J.C.; Franklin, B.A. Exercise Preconditioning as a Cardioprotective Phenotype. Am. J. Cardiol. 2021, 148, 8–15. [Google Scholar] [CrossRef]
- Burelle, Y.; Wambolt, R.B.; Grist, M.; Parsons, H.L.; Chow, J.C.; Antler, C.; Bonen, A.; Keller, A.; Dunaway, G.A.; Popov, K.M.; et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am. J. Physiology. Heart Circ. Physiol. 2004, 287, H1055–H1063. [Google Scholar] [CrossRef]
- Oi, Y.; Nagoshi, T.; Kimura, H.; Tanaka, Y.; Yoshii, A.; Yasutake, R.; Takahashi, H.; Kashiwagi, Y.; Tanaka, T.D.; Tachibana, T.; et al. Exogenous ANP Treatment Ameliorates Myocardial Insulin Resistance and Protects against Ischemia-Reperfusion Injury in Diet-Induced Obesity. Int. J. Mol. Sci. 2022, 23, 8373. [Google Scholar] [CrossRef]
- Lee, Y.; Min, K.; Talbert, E.E.; Kavazis, A.N.; Smuder, A.J.; Willis, W.T.; Powers, S.K. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med. Sci. Sport. Exerc. 2012, 44, 397–405. [Google Scholar] [CrossRef]
- Sarzani, R.; Allevi, M.; Di Pentima, C.; Schiavi, P.; Spannella, F.; Giulietti, F. Role of Cardiac Natriuretic Peptides in Heart Structure and Function. Int. J. Mol. Sci. 2022, 23, 14415. [Google Scholar] [CrossRef]
- Numata, G.; Takimoto, E. Cyclic GMP and PKG Signaling in Heart Failure. Front. Pharm. 2022, 13, 792798. [Google Scholar] [CrossRef]
- Gudi, T.; Casteel, D.E.; Vinson, C.; Boss, G.R.; Pilz, R.B. NO activation of fos promoter elements requires nuclear translocation of G-kinase I and CREB phosphorylation but is independent of MAP kinase activation. Oncogene 2000, 19, 6324–6333. [Google Scholar] [CrossRef] [Green Version]
- Pilz, R.B.; Broderick, K.E. Role of cyclic GMP in gene regulation. Front. Biosci. 2005, 10, 1239–1268. [Google Scholar] [CrossRef] [Green Version]
- Domondon, M.; Nikiforova, A.B.; DeLeon-Pennell, K.Y.; Ilatovskaya, D.V. Regulation of mitochondria function by natriuretic peptides. Am. J. Physiol. Ren. Physiol. 2019, 317, F1164–F1168. [Google Scholar] [CrossRef]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef]
- Colbran, J.L.; Roach, P.J.; Fiol, C.J.; Dixon, J.E.; Andrisani, O.M.; Corbin, J.D. cAMP-dependent protein kinase, but not the cGMP-dependent enzyme, rapidly phosphorylates delta-CREB, and a synthetic delta-CREB peptide. Biochem. Cell Biol. 1992, 70, 1277–1282. [Google Scholar] [CrossRef]
- Servey, J.T.; Stephens, M. Cardiac Rehabilitation: Improving Function and Reducing Risk. Am. Fam. Physician 2016, 94, 37–43. [Google Scholar]
- Working Group on Cardiac Rehabilitation & Exercise Physiology; Working Group on Heart Failure of the European Society of Cardiology. Recommendations for exercise training in chronic heart failure patients. Eur. Heart J. 2001, 22, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Chicco, A.J.; Jew, K.N.; Johnson, M.S.; Lynch, J.M.; Watson, P.A.; Moore, R.L. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J. Physiol. 2005, 569, 913. [Google Scholar] [CrossRef]
- Demirel, H.A.; Powers, S.K.; Zergeroglu, M.A.; Shanely, R.A.; Hamilton, K.; Coombes, J.; Naito, H. Short-term exercise improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. J. Appl. Physiol. 2001, 91, 2205–2212. [Google Scholar] [CrossRef]
- French, J.P.; Quindry, J.C.; Falk, D.J.; Staib, J.L.; Lee, Y.; Wang, K.K.; Powers, S.K. Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am. J. Physiology. Heart Circ. Physiol. 2006, 290, H128–H136. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, K.L.; Quindry, J.C.; French, J.P.; Staib, J.; Hughes, J.; Mehta, J.L.; Powers, S.K. MnSOD antisense treatment and exercise-induced protection against arrhythmias. Free. Radic. Biol. Med. 2004, 37, 1360–1368. [Google Scholar] [CrossRef]
- Hamilton, K.L.; Staib, J.L.; Phillips, T.; Hess, A.; Lennon, S.L.; Powers, S.K. Exercise, antioxidants, and HSP72: Protection against myocardial ischemia/reperfusion. Free. Radic. Biol. Med. 2003, 34, 800–809. [Google Scholar] [CrossRef]
- Hamilton, K.L.; Powers, S.K.; Sugiura, T.; Kim, S.; Lennon, S.; Tumer, N.; Mehta, J.L. Short-term exercise training can improve myocardial tolerance to I/R without elevation in heat shock proteins. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1346–H1352. [Google Scholar] [CrossRef]
- Lennon, S.L.; Quindry, J.; Hamilton, K.L.; French, J.; Staib, J.; Mehta, J.L.; Powers, S.K. Loss of exercise-induced cardioprotection after cessation of exercise. J. Appl. Physiol. 2004, 96, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Lennon, S.L.; Quindry, J.C.; Hamilton, K.L.; French, J.P.; Hughes, J.; Mehta, J.L.; Powers, S.K. Elevated MnSOD is not required for exercise-induced cardioprotection against myocardial stunning. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H975–H980. [Google Scholar] [CrossRef] [Green Version]
- Libonati, J.R.; Kendrick, Z.V.; Houser, S.R. Sprint training improves postischemic, left ventricular diastolic performance. J. Appl. Physiol. 2005, 99, 2121–2127. [Google Scholar] [CrossRef] [Green Version]
- Libonati, J.R. Exercise training improves left ventricular isovolumic relaxation. Med. Sci. Sport. Exerc. 2000, 32, 1399–1405. [Google Scholar] [CrossRef]
- Libonati, J.R.; Gaughan, J.P.; Hefner, C.A.; Gow, A.; Paolone, A.M.; Houser, S.R. Reduced ischemia and reperfusion injury following exercise training. Med. Sci. Sport. Exerc. 1997, 29, 509–516. [Google Scholar] [CrossRef]
- Powers, S.K.; Demirel, H.A.; Vincent, H.K.; Coombes, J.S.; Naito, H.; Hamilton, K.L.; Shanely, R.A.; Jessup, J. Exercise training improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. Am. J. Physiol. 1998, 275, R1468–R1477. [Google Scholar] [CrossRef]
- Quindry, J.; French, J.; Hamilton, K.; Lee, Y.; Mehta, J.L.; Powers, S. Exercise training provides cardioprotection against ischemia-reperfusion induced apoptosis in young and old animals. Exp. Gerontol. 2005, 40, 416–425. [Google Scholar] [CrossRef]
- Starnes, J.W.; Taylor, R.P.; Park, Y. Exercise improves postischemic function in aging hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H347–H351. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.P.; Harris, M.B.; Starnes, J.W. Acute exercise can improve cardioprotection without increasing heat shock protein content. Am. J. Physiol. 1999, 276, H1098–H1102. [Google Scholar] [CrossRef]
- Yamashita, N.; Hoshida, S.; Otsu, K.; Asahi, M.; Kuzuya, T.; Hori, M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J. Exp. Med. 1999, 189, 1699–1706. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, S.; Yamashita, N.; Otsu, K.; Hori, M. Repeated physiologic stresses provide persistent cardioprotection against ischemia-reperfusion injury in rats. J. Am. Coll. Cardiol. 2002, 40, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Quindry, J.; Hamilton, K. Aging, exercise, and cardioprotection. Ann. N. Y. Acad. Sci. 2004, 1019, 462–470. [Google Scholar] [CrossRef]
- Powers, S.K.; Locke; Demirel, H.A. Exercise, heat shock proteins, and myocardial protection from I-R injury. Med. Sci. Sport. Exerc. 2001, 33, 386–392. [Google Scholar] [CrossRef]
- Ascensao, A.; Ferreira, R.; Magalhaes, J. Exercise-induced cardioprotection—Biochemical, morphological and functional evidence in whole tissue and isolated mitochondria. Int. J. Cardiol. 2007, 117, 16–30. [Google Scholar] [CrossRef]
- Powers, S.K.; Quindry, J.C.; Kavazis, A.N. Exercise-induced cardioprotection against myocardial ischemia-reperfusion injury. Free Radic. Biol. Med. 2008, 44, 193–201. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide and mitochondria. Front. Biosci. 2007, 12, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar] [CrossRef] [Green Version]
- Seo, D.Y.; Kwak, H.B.; Kim, A.H.; Park, S.H.; Heo, J.W.; Kim, H.K.; Ko, J.R.; Lee, S.J.; Bang, H.S.; Sim, J.W.; et al. Cardiac adaptation to exercise training in health and disease. Pflug. Arch. 2020, 472, 155–168. [Google Scholar] [CrossRef]
- Tao, L.; Bei, Y.; Lin, S.; Zhang, H.; Zhou, Y.; Jiang, J.; Chen, P.; Shen, S.; Xiao, J.; Li, X. Exercise Training Protects Against Acute Myocardial Infarction via Improving Myocardial Energy Metabolism and Mitochondrial Biogenesis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 162–175. [Google Scholar] [CrossRef]
- Wang, H.; Bei, Y.; Lu, Y.; Sun, W.; Liu, Q.; Wang, Y.; Cao, Y.; Chen, P.; Xiao, J.; Kong, X. Exercise Prevents Cardiac Injury and Improves Mitochondrial Biogenesis in Advanced Diabetic Cardiomyopathy with PGC-1alpha and Akt Activation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 35, 2159–2168. [Google Scholar] [CrossRef]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: Role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E519–E528. [Google Scholar] [CrossRef] [Green Version]
- Ciani, E.; Guidi, S.; Bartesaghi, R.; Contestabile, A. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: Implication for a survival role of nitric oxide. J. Neurochem. 2002, 82, 1282–1289. [Google Scholar] [CrossRef]
- Wu, G.Y.; Deisseroth, K.; Tsien, R.W. Activity-dependent CREB phosphorylation: Convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 2808–2813. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Hu, C.; Yuan, X.P.; Yuan, Y.P.; Song, P.; Kong, C.Y.; Teng, T.; Hu, M.; Xu, S.C.; Ma, Z.G.; et al. Osteocrin, a novel myokine, prevents diabetic cardiomyopathy via restoring proteasomal activity. Cell. Death Dis. 2021, 12, 624. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, X.; Zhang, N.; Wei, W.Y.; Li, L.L.; Ma, Z.G.; Tang, Q.Z. Osteocrin attenuates inflammation, oxidative stress, apoptosis, and cardiac dysfunction in doxorubicin-induced cardiotoxicity. Clin. Transl. Med. 2020, 10, e124. [Google Scholar] [CrossRef]
- Drummond, M.J.; Dreyer, H.C.; Fry, C.S.; Glynn, E.L.; Rasmussen, B.B. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J. Appl. Physiol. 2009, 106, 1374–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, L.R. Natriuretic peptide metabolism, clearance and degradation. FEBS J. 2011, 278, 1808–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Wu, Z.; Li, Z. Efficacy and safety of nesiritide in patients with decompensated heart failure: A meta-analysis of randomised trials. BMJ Open 2016, 6, e008545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sa, D.D.; Chen, H.H. The role of natriuretic peptides in heart failure. Curr. Cardiol. Rep. 2008, 10, 182–189. [Google Scholar] [CrossRef]
- Takaya, Y.; Yoshihara, F.; Yokoyama, H.; Kanzaki, H.; Kitakaze, M.; Goto, Y.; Anzai, T.; Yasuda, S.; Ogawa, H.; Kawano, Y.; et al. Impact of decreased serum albumin levels on acute kidney injury in patients with acute decompensated heart failure: A potential association of atrial natriuretic peptide. Heart Vessel. 2017, 32, 932–943. [Google Scholar] [CrossRef]
- Moyes, A.J.; Khambata, R.S.; Villar, I.; Bubb, K.J.; Baliga, R.S.; Lumsden, N.G.; Xiao, F.; Gane, P.J.; Rebstock, A.S.; Worthington, R.J.; et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J. Clin. Investig. 2014, 124, 4039–4051. [Google Scholar] [CrossRef]
- Moyes, A.J.; Chu, S.M.; Aubdool, A.A.; Dukinfield, M.S.; Margulies, K.B.; Bedi, K.C.; Hodivala-Dilke, K.; Baliga, R.S.; Hobbs, A.J. C-type natriuretic peptide co-ordinates cardiac structure and function. Eur. Heart J. 2020, 41, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Bubb, K.J.; Aubdool, A.A.; Moyes, A.J.; Lewis, S.; Drayton, J.P.; Tang, O.; Mehta, V.; Zachary, I.C.; Abraham, D.J.; Tsui, J.; et al. Endothelial C-Type Natriuretic Peptide Is a Critical Regulator of Angiogenesis and Vascular Remodeling. Circulation 2019, 139, 1612–1628. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engeli, S.; Birkenfeld, A.L.; Badin, P.M.; Bourlier, V.; Louche, K.; Viguerie, N.; Thalamas, C.; Montastier, E.; Larrouy, D.; Harant, I.; et al. Natriuretic peptides enhance the oxidative capacity of human skeletal muscle. J. Clin. Investig. 2012, 122, 4675–4679. [Google Scholar] [CrossRef] [Green Version]
- Zois, N.E.; Bartels, E.D.; Hunter, I.; Kousholt, B.S.; Olsen, L.H.; Goetze, J.P. Natriuretic peptides in cardiometabolic regulation and disease. Nat. Reviews. Cardiol. 2014, 11, 403–412. [Google Scholar] [CrossRef]
- Hamasaki, H. The Effects of Exercise on Natriuretic Peptides in Individuals without Heart Failure. Sports 2016, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Kerkela, R.; Ulvila, J.; Magga, J. Natriuretic Peptides in the Regulation of Cardiovascular Physiology and Metabolic Events. J. Am. Heart Assoc. 2015, 4, e002423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruden, G.; Landi, A.; Bruno, G. Natriuretic peptides, heart, and adipose tissue: New findings and future developments for diabetes research. Diabetes Care 2014, 37, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Lee, W.; Arrowsmith, C.H.; Muhandiram, D.R.; Kay, L.E. A suite of triple-resonance NMR experiments for the backbone assignment of 15N, 13C, 2H-labeled proteins with high sensitivity. J. Am. Chem. Soc. 1994, 116, 11655–11666. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M. Multidimensional heteronuclear magnetic resonance of proteins. Meths. Enzymol. 1994, 239, 349–363. [Google Scholar]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Johnson, B.A.; Blevins, R.A. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.P.; Zhang, Q.J.; Cochran, C.T.; Ponce, J.; Alexander, S.; Kronemberger, A.; Fuqua, J.D.; Zhang, Y.; Fattal, R.; Harper, T.; et al. Perinatal versus adult loss of ULK1 and ULK2 distinctly influences cardiac autophagy and function. Autophagy 2022, 18, 2161–2177. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harris, M.P.; Zeng, S.; Zhu, Z.; Lira, V.A.; Yu, L.; Hodgson-Zingman, D.M.; Zingman, L.V. Myokine Musclin Is Critical for Exercise-Induced Cardiac Conditioning. Int. J. Mol. Sci. 2023, 24, 6525. https://doi.org/10.3390/ijms24076525
Harris MP, Zeng S, Zhu Z, Lira VA, Yu L, Hodgson-Zingman DM, Zingman LV. Myokine Musclin Is Critical for Exercise-Induced Cardiac Conditioning. International Journal of Molecular Sciences. 2023; 24(7):6525. https://doi.org/10.3390/ijms24076525
Chicago/Turabian StyleHarris, Matthew P., Shemin Zeng, Zhiyong Zhu, Vitor A. Lira, Liping Yu, Denice M. Hodgson-Zingman, and Leonid V. Zingman. 2023. "Myokine Musclin Is Critical for Exercise-Induced Cardiac Conditioning" International Journal of Molecular Sciences 24, no. 7: 6525. https://doi.org/10.3390/ijms24076525