

CAR T Cell Therapy: A Versatile Living Drug

Abstract
:1. Introduction
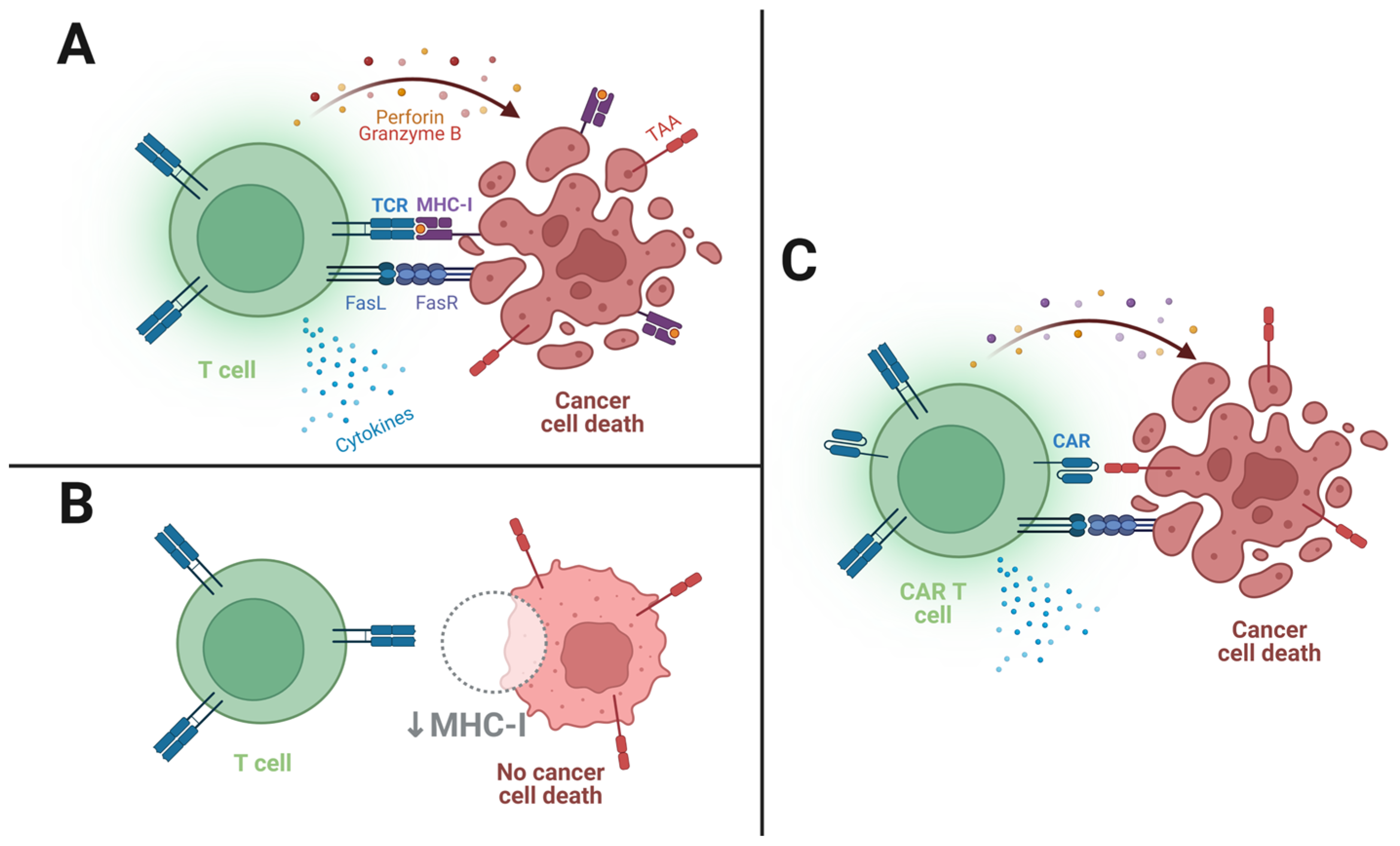
2. How Do CAR T Cells Kill?
3. Currently Available CAR T Cell Therapies
- CD19 expression is both quite limited to, and ubiquitous in, B cells [23]; therefore, its targeting avoids toxicity to other tissues while assuring the targeting of malignant B cells;
- As the anti-CD19 CAR T cells target all B cells in the patient’s body, the therapies will frequently cause B cell aplasia and consequent hypogammaglobulinemia (low serum antibody levels) [24]. Fortunately, although debilitating as they may be, these conditions are compatible with resuming a relatively normal life, especially with the use of immunoglobulin infusions [25].
Current Status
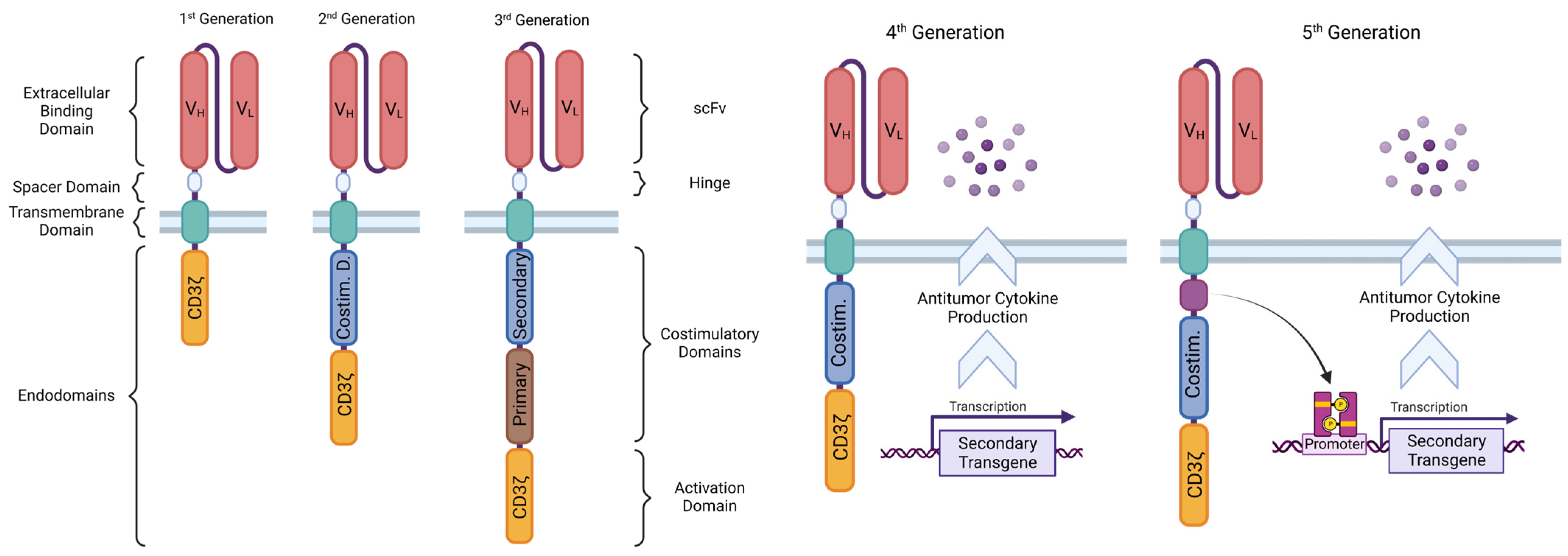
4. Structure of the CAR
4.1. Ligand-Binding Domain
4.1.1. scFv
4.1.2. Other LBDs
4.2. Spacer Domain
4.3. Transmembrane Domain
4.4. Endodomains
4.4.1. CD3ζ Signaling Domain
4.4.2. Costimulatory Domains
- CD28 heightens the signaling speed and intensity and cytokine production, induces effector memory differentiation on the activated CAR T cells, and reduces effects of inhibitory molecules and Tregs, increasing the CAR T cells’ cytolytic and inflammatory potential; however, it imparts a tendency for fast exhaustion, and thus lower persistence, than 4-1BB-containing second-generation products [55]. The more “explosive” behavior of CD28 CAR T cells seems to also bring along a downside of greater chances for and intensity of CRS and neurological toxicity [62,63].
4.5. CAR T Generations According to Structure
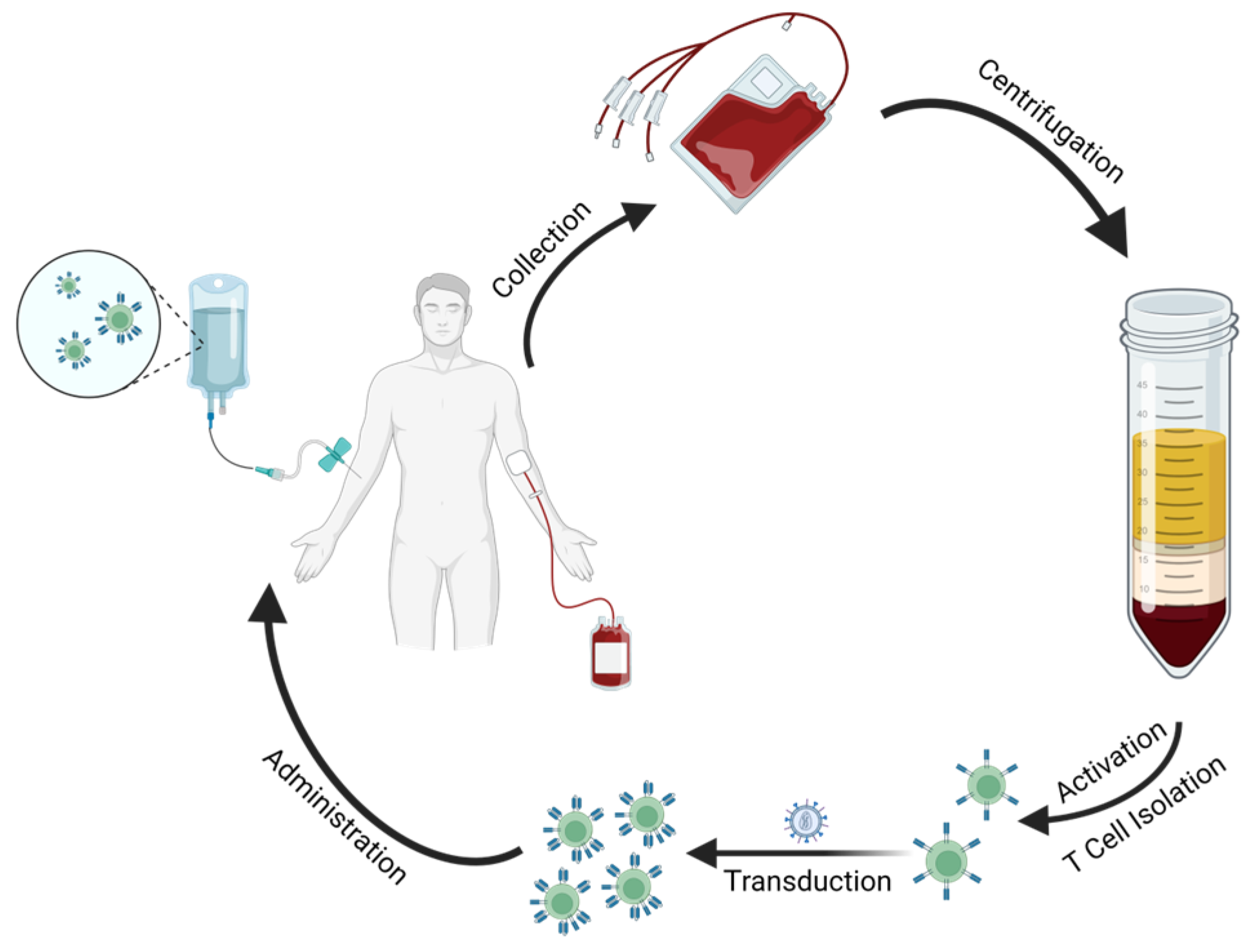
5. CAR T Cell Processing
5.1. Manufacturing
5.2. Administration
6. Current Challenges
6.1. Efficacy and Persistence
6.1.1. Tumor-Antigen Escape
6.1.2. Solid Tumors
- First and foremost, CAR T cells are infused into the blood, and must then traffic to the region where the tumor is located, a process which is dependent on chemokine attraction signals and is therefore variable from tumor to tumor [75].
- After reaching the tumor site, the lymphocytes must penetrate through the layers of ECM, frequently thickened and stiffened by intense collagen and heparan sulphate proteoglycan deposition carried out by tumor-associated fibroblasts. To make matters worse, T cells do not produce significant amounts of ECM-degrading enzymes, meaning they tend to move less through areas with a denser matrix. Hence, this barrier hinders deeply the accessibility of CAR T cells to their target cells [77].
- Atop this, the microenvironment of solid tumors is often oxidative, hypoxic, acidic, and nutrient-starved, and consists of high amounts of immunosuppressive elements, be they cytokines and other soluble factors, cells (e.g., Tregs, tumor-associated macrophages (TAMs) and MDSCs), or even the tumor cells themselves, through the expression of ligands like CTLA-4 and PD-L1. This immunologically deleterious “cold tumor” environment spurs the development of anergic and apoptotic states in the CAR T cells [78].
- Unlike B cell malignancies, it has proven challenging to find TAAs that are specifically yet uniformly expressed in the tumor at high levels. TAAs can be found that generally present higher expression in cancer cells. However, they are, more often than not, also coexpressed in low levels in non-malignant tissues, enabling dangerous cross-reactivity and on-target off-tumor toxicity [75,79,80]. This also means that CAR T cell therapies that end up proceeding to clinical trials, although deemed safe enough in preclinical testing, often spur cases of severe and sometimes deadly toxicity, leading to product failure [76,81]. Even when dealing with TAAs of very low expression in healthy cells, solid tumors are usually very heterogeneous, so wide antigen expression variability and antigen-loss events are quite common [80].
6.2. Safety
6.2.1. Cytokine-Release Syndrome
6.2.2. Neurotoxicity
6.2.3. On-Target Off-Tumor Toxicity
- normal ones, with no expression of the specific antigen, that go by immunologically invisible.
- target malignant cells, which highly express the antigen and are therefore attacked by the lymphocytes.
- problematic, target antigen-negative malignant cells, which also often escape unscathed from the cytolytic action of CAR T cells.
- normal cells expressing the target antigen, which, unfortunately, get caught in the immune crossfire and end up succumbing to the inflammatory and cytolytic action of the CAR T cells—this “friendly-fire” can have serious consequences, damaging healthy tissue and compromising its function, besides creating unnecessary inflammation, which can have detrimental effects both locally and systemically [87].
6.2.4. Other Safety Concerns
7. Innovative Research
7.1. Special CAR T Engineering
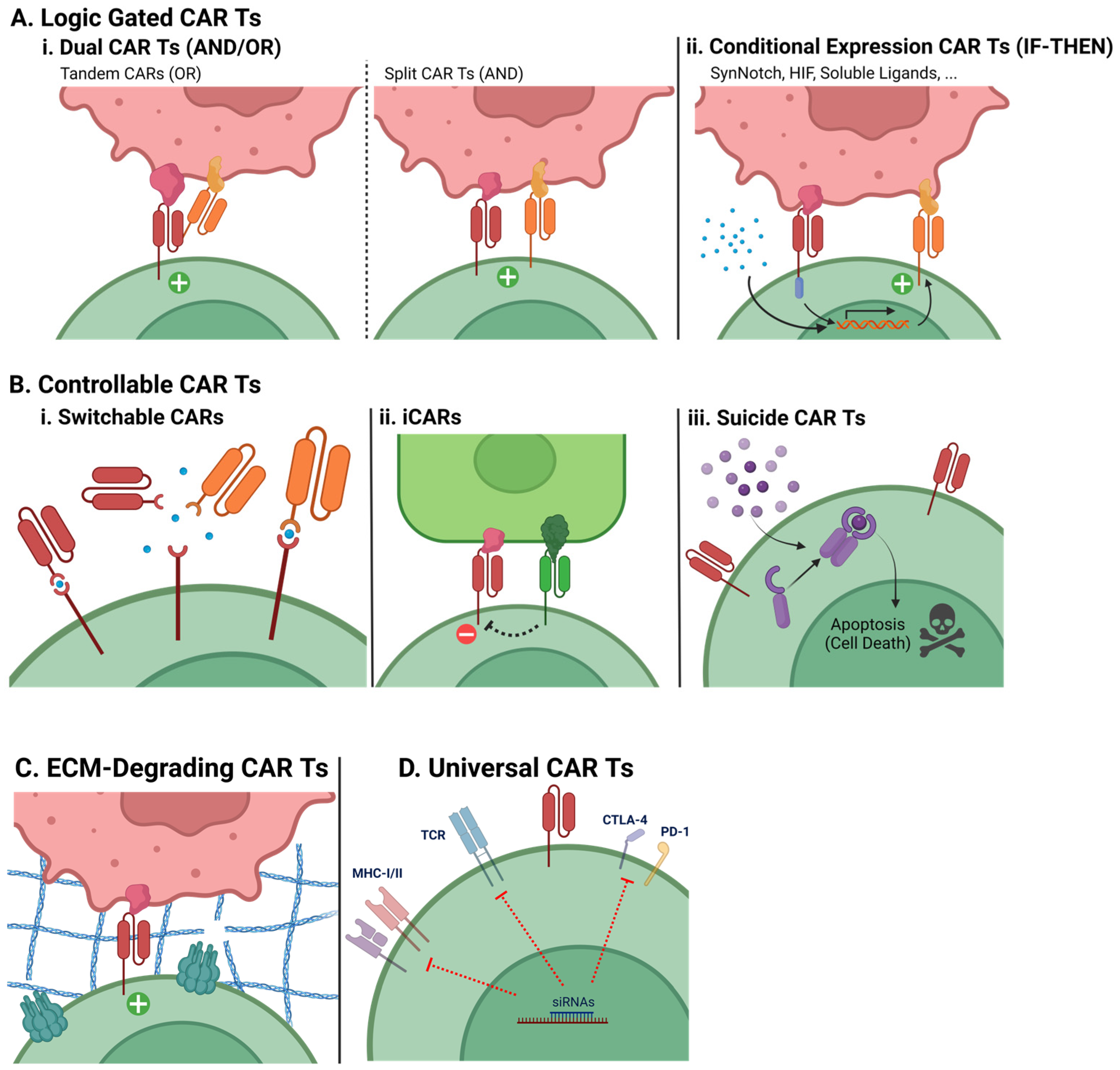
7.1.1. Logic-Gated Strategies
- Dual CAR T cells (AND/OR)
- Conditional Expression CAR T cells (IF-THEN)
7.1.2. Controllable CAR T cells
- Switchable CARs
- Inhibitory CARs
- Suicide CAR T cells
7.1.3. ECM-Degrading CAR T Cells
- Universal CAR T cells
7.2. CAR T Cell Therapy beyond Cancer
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Um, P. Cancer, Definition. In Encyclopedia of Metagenomics; Highlander, S.K., Rodriguez-Valera, F., White, B.A., Eds.; Springer: Boston, MA, USA, 2015; p. 65. ISBN 978-1-4899-7475-4. [Google Scholar]
- Carbone, A. Cancer Classification at the Crossroads. Cancers 2020, 12, 980. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salk, J.J.; Fox, E.J.; Loeb, L.A. Mutational Heterogeneity in Human Cancers. Annu. Rev. Pathol. 2010, 5, 51–75. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Sun, X.X.; Yu, Q. Intra-Tumor Heterogeneity of Cancer Cells and Its Implications for Cancer Treatment. Acta Pharmacol. Sin. 2015, 36, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor Heterogeneity: Causes and Consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, Á. Assessment of the Evolution of Cancer Treatment Therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Liu, Z.; Fu, Y.X. Adapting Conventional Cancer Treatment for Immunotherapy. J. Mol. Med. 2016, 94, 489–495. [Google Scholar] [CrossRef]
- Wu, H.-C.; Chang, D.-K.; Huang, C.-T. Targeted Therapy to Treat Cancer. J. Cancer Mol. 2006, 2, 57–66. [Google Scholar]
- Li, Y.; Ayala-Orozco, C.; Rauta, P.R.; Krishnan, S. The Application of Nanotechnology in Enhancing Immunotherapy for Cancer Treatment: Current Effects and Perspective. Nanoscale 2019, 11, 17157–17178. [Google Scholar] [CrossRef]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J.B.A.G. Adoptive Cellular Therapies: The Current Landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther.—Methods Clin. Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.H.; Fiorenza, S.; Koldej, R.M.; Jaworowski, A.; Ritchie, D.S.; Quinn, K.M. T Cell Fitness and Autologous CAR T Cell Therapy in Haematologic Malignancy. Front. Immunol. 2021, 12, 780442. [Google Scholar] [CrossRef]
- Chang, Z.L.; Chen, Y.Y. CARs: Synthetic Immunoreceptors for Cancer Therapy and Beyond. Trends Mol. Med. 2017, 23, 430–450. [Google Scholar] [CrossRef] [Green Version]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, 2–10. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric Antigen Receptor T Cells Form Nonclassical and Potent Immune Synapses Driving Rapid Cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [Green Version]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [Green Version]
- Ivica, N.A.; Young, C.M. Tracking the Car-t Revolution: Analysis of Clinical Trials of Car-t and Tcr-t Therapies for the Treatment of Cancer (1997–2020). Healthcare 2021, 9, 1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A Biomarker for B Cell Development, Lymphoma Diagnosis and Therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wat, J.; Barmettler, S. Hypogammaglobulinemia After Chimeric Antigen Receptor (CAR) T-Cell Therapy: Characteristics, Management, and Future Directions. J. Allergy Clin. Immunol. Pract. 2022, 10, 460–466. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Jarrell, D.K.; Drake, S.; Brown, M.A. Advancing Therapies for Cancer—From Mustard Gas to CAR T. Science 2020, 2, 90. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.T.; Carpenter, R.O.; Maryalice, S.S.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated with Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, e321–e327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 Expression in T Cells for Effective Chimeric Antigen Receptor Targeting of T-Cell Malignancies. Blood Adv. 2017, 1, 2348–2360. [Google Scholar] [CrossRef] [Green Version]
- Halim, L.; Maher, J. CAR T-Cell Immunotherapy of B-Cell Malignancy: The Story so Far. Ther. Adv. Vaccines Immunother. 2020, 8, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Yescarta®. CAR T-Cell Therapy Demonstrates Consistent Survival Outcomes and Safety in Real-World Setting Regardless of Race and Ethnicity. Available online: https://www.businesswire.com/news/home/20220603005447/en/ (accessed on 8 March 2023).
- Yescarta®. Is First CAR T-Cell Therapy to Report Five-Year Survival Data from Pivotal Study Showing Durable Long-Term Survival in Patients with Refractory Large B-Cell Lymphoma. Available online: https://www.businesswire.com/news/home/20211211005050/en/ (accessed on 8 March 2023).
- Golden, D. Kymriah’s 5-Year Survival Data Shows Promise of CAR T-Cell Therapy. Available online: https://acgtfoundation.org/news/kymriah-survival-data-shows-promise-of-car-t-cell-therapy/ (accessed on 8 March 2023).
- New Analyses of Kite’s Tecartus® CAR T-Cell Therapy Provide Additional Evidence Supporting Overall Survival and Durability of Response. Available online: https://www.businesswire.com/news/home/20221212005230/en/ (accessed on 8 March 2023).
- Hansen, D.K.; Sidana, S.; Peres, L.C.; Colin Leitzinger, C.; Shune, L.; Shrewsbury, A.; Gonzalez, R.; Sborov, D.W.; Wagner, C.; Dima, D.; et al. Idecabtagene Vicleucel for Relapsed/Refractory Multiple Myeloma: Real-World Experience From the Myeloma CAR T Consortium. J. Clin. Oncol. 2023, 22, 01365. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Dekervel, J.; Hendlisz, A.; Anguille, S.; Awada, A.; Cerf, E.; Lonez, C.; Breman, E.; Dheur, M.-S.; Alcantar-Orozco, E.; et al. Updated Data from AlloSHRINK Phase I First-in-Human Study Evaluating CYAD-101, an Innovative Non-Gene Edited Allogeneic CAR-T in MCRC. J. Clin. Oncol. 2021, 39, 74. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I Study of Chimeric Antigen Receptor Modified T Cells in Treating HER2-Positive Advanced Biliary Tract Cancers and Pancreatic Cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef]
- Olson, M.L.; Mause, E.R.V.; Radhakrishnan, S.V.; Brody, J.D.; Rapoport, A.P.; Welm, A.L.; Atanackovic, D.; Luetkens, T. Low-Affinity CAR T Cells Exhibit Reduced Trogocytosis, Preventing Rapid Antigen Loss, and Increasing CAR T Cell Expansion. Leukemia 2022, 36, 1943–1946. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Chowdhury, A.; Karulkar, A.; Jaiswal, A.K.; Banik, A.; Asija, S.; Purwar, R. Immunogenicity of CAR-T Cell Therapeutics: Evidence, Mechanism and Mitigation. Front. Immunol. 2022, 13, 886546. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-Independent Activation Enhances the Efficacy of 4-1BB-Costimulated CD22 CAR T Cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, B.; Cook, W.J.; Murad, J.; Graber, D.J.; Sentman, M.L.; Lonez, C.; Gilham, D.E.; Sentman, C.L.; Agaugue, S. Exploiting Natural Killer Group 2D Receptors for CAR T-Cell Therapy. Futur. Oncol. 2017, 13, 1593–1605. [Google Scholar] [CrossRef] [Green Version]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-Cells Forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yu, L.; Cooper, L.J.N.; Hollomon, M.; Huls, H.; Kleinerman, E.S. Genetically Modified T Cells Targeting Interleukin-11 Receptor α-Chain Kill Human Osteosarcoma Cells and Induce the Regression of Established Osteosarcoma Lung Metastases. Cancer Res. 2012, 72, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Gao, Q.; Zhou, K.; Zhou, J.; Fang, B.; Zhang, J.; Li, H.; Song, Y. The Phase I Clinical Study of CART Targeting BCMA with Humanized Alpaca-Derived Single-Domain Antibody as Antigen Recognition Domain. J. Clin. Oncol. 2019, 37, 2535. [Google Scholar] [CrossRef]
- Siegler, E.; Li, S.; Kim, Y.J.; Wang, P. Designed Ankyrin Repeat Proteins as Her2 Targeting Domains in Chimeric Antigen Receptor-Engineered T Cells. Hum. Gene Ther. 2017, 28, 726–736. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of Human T Cells to Tumor-Associated MUC1: The Evolution of a Chimeric Antigen Receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T Cell Persistence through ICOS and 4-1BB Costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Gil, D.; Schamel, W.W.A.; Montoya, M.; Sánchez-Madrid, F.; Alarcón, B. Recruitment of Nck by CD3ε Reveals a Ligand- Induced Conformational Change Essential for T Cell Receptor Signaling and Synapse Formation. Cell 2002, 109, 901–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.S.; Glassman, C.R.; Deshpande, N.R.; Badgandi, H.B.; Parrish, H.L.; Uttamapinant, C.; Stawski, P.S.; Ting, A.Y.; Kuhns, M.S. A Mechanical Switch Couples T Cell Receptor Triggering to the Cytoplasmic Juxtamembrane Regions of CD3ζζ. Immunity 2015, 43, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.; Al-Haidari, A.; Sun, J.; Kazi, J.U. T Cell Receptor (TCR) Signaling in Health and Disease. Signal Transduct. Target. Ther. 2021, 6, 412. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.H.; de Jong, A.; Jukema, J.W.; de Vries, M.R.; Arens, R.; Quax, P.H.A. T Cell Co-Stimulation and Co-Inhibition in Cardiovascular Disease: A Double-Edged Sword. Nat. Rev. Cardiol. 2019, 16, 325–343. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. A Comparison of Chimeric Antigen Receptors Containing CD28 versus 4-1BB Costimulatory Domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther.–Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific Activation and Targeting of Cytotoxic Lymphocytes through Chimeric Single Chains Consisting of Antibody-Binding Domains and the γ or ζ Subunits of the Immunoglobulin and T-Cell Receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (Tisagenlecleucel): CAR-T Therapy for Relapsed and Refractory B-Cell Acute Lymphoblastic Leukemia. Drug Des. Dev. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [Green Version]
- Casati, A.; Varghaei-Nahvi, A.; Feldman, S.A.; Assenmacher, M.; Rosenberg, S.A.; Dudley, M.E.; Scheffold, A. Clinical-Scale Selection and Viral Transduction of Human Naïve and Central Memory CD8+ T Cells for Adoptive Cell Therapy of Cancer Patients. Cancer Immunol. Immunother. 2013, 62, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Mo, F.; McKenna, M.K. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front. Immunol. 2022, 13, 876339. [Google Scholar] [CrossRef]
- Lukjanov, V.; Koutná, I.; Šimara, P. CAR T-Cell Production Using Nonviral Approaches. J. Immunol. Res. 2021, 2021, 6644685. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated with High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Amini, L.; Silbert, S.K.; Maude, S.L.; Nastoupil, L.J.; Ramos, C.A.; Brentjens, R.J.; Sauter, C.S.; Shah, N.N.; Abou-el-Enein, M. Preparing for CAR T Cell Therapy: Patient Selection, Bridging Therapies and Lymphodepletion. Nat. Rev. Clin. Oncol. 2022, 19, 342–355. [Google Scholar] [CrossRef]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Susanibar-Adaniya, S.; Barta, S.K. 2021 Update on Diffuse Large B Cell Lymphoma: A Review of Current Data and Potential Applications on Risk Stratification and Management. Am. J. Hematol. 2021, 96, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical Development of CAR T Cells—Challenges and Opportunities in Translating Innovative Treatment Concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Wang, L.C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells Can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T Cell Therapy and the Tumor Microenvironment: Current Challenges and Opportunities. Mol. Ther.–Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-mora, F.; Gallego-ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic. Cells 2020, 27, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming On-Target, off-Tumour Toxicity of CAR T Cell Therapy for Solid Tumours. Nat. Rev. Clin. Oncol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Si, S.; Teachey, D.T. Spotlight on Tocilizumab in the Treatment of Car-t-Cell-Induced Cytokine Release Syndrome: Clinical Evidence to Date. Ther. Clin. Risk Manag. 2020, 16, 705–714. [Google Scholar] [PubMed]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine Release Syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of Chimeric Antigen Receptor T Cells: Recognition and Management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with Car T-Cell Therapy in Patients with B-Cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-Modified T Cells: Toxicities and Overcoming Strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef]
- García-Guerrero, E.; Sierro-Martínez, B.; Pérez-Simón, J.A. Overcoming Chimeric Antigen Receptor (CAR) Modified T-Cell Therapy Limitations in Multiple Myeloma. Front. Immunol. 2020, 11, 1128. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and Management in CAR T-Cell Therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Blache, U.; Popp, G.; Dünkel, A.; Koehl, U.; Fricke, S. Potential Solutions for Manufacture of CAR T Cells in Cancer Immunotherapy. Nat. Commun. 2022, 13, 5225. [Google Scholar] [CrossRef]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.W.C.J.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- van der Schans, J.J.; van de Donk, N.W.C.J.; Mutis, T. Dual Targeting to Overcome Current Challenges in Multiple Myeloma CAR T-Cell Treatment. Front. Oncol. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T Cells Overcome Challenges of Specificity, Heterogeneity, and Persistence in Treating Glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-Mediated Activation and Retargeting of CAR-T Cells for B-Cell Malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, A.R.; Owens, M.N.; Geyer, C.R. Modular Chimeric Antigen Receptor Systems for Universal CAR T Cell Retargeting. Int. J. Mol. Sci. 2020, 21, 7222. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-Antigen-Targeted Chimeric Antigen Receptor T Cells for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorenza, S.; Ritchie, D.S.; Ramsey, S.D.; Turtle, C.J.; Roth, J.A. Value and Affordability of CAR T-Cell Therapy in the United States. Bone Marrow Transpl. 2020, 55, 1706–1715. [Google Scholar] [CrossRef]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T Cells in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T Cells to Kill Cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T Cell Therapy for Refractory Systemic Lupus Erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Commercial Name | Product Name | Manufacturer | Application | Approval |
|---|---|---|---|---|
| Yescarta | Axicabtagene ciloleucel (anti-CD19) | Kite Pharma, Inc. (Los Angeles, CA, USA) | LBCL | EMA and FDA |
| HGBCL | FDA | |||
| PMBCL | EMA and FDA | |||
| FL | EMA and FDA | |||
| Kymriah | Tisagenlecleucel (anti-CD19) | Novartis Pharmaceutical Corporation (Basel, Switzerland) | LBCL | EMA and FDA |
| HGBCL | FDA | |||
| FL | EMA and FDA | |||
| B-ALL | EMA and FDA | |||
| Breyanzi | Lisocabtagene maraleucel (anti-CD19) | Juno Therapeutics, Inc. (Bristol-Meyers Squibb) (Seattle, WA, USA) | LBCL | EMA and FDA |
| HGBCL | FDA | |||
| PMBCL | EMA and FDA | |||
| FL3B | EMA and FDA | |||
| Tecartus | Brexucabtagene autoleucel (anti-CD19) | Kite Pharma, Inc. (Los Angeles, CA, USA) | MCL | EMA and FDA |
| B-ALL | FDA | |||
| Abecma | Idecabtagene vicleucel (anti-BCMA) | Celgene Corporation (Bristol-Meyers Squibb) (Summit, NJ, USA) | MM | EMA and FDA |
| Carvykti | Ciltacabtagene autoleucel (anti-BCMA) | Janssen Biotech, Inc. (Beerse, Belgium) | MM | EMA and FDA |
| CAR T | Cancer Type | Results | Status |
|---|---|---|---|
| CD19-targeting CARs (Yescarta, Kymriah, Breyanzi, Tecartus) | Various lymphomas and leukemias | Yescarta: overall survival at 12 months of 65%, 42.6% at 5 years [37,38]. Kymriah: 82% remission rate, 5-year relapse-free survival rate of 44% and 5-year overall survival rate of 55% [39]. Breyanzi: event- and progression-free survival of 10.1 months and 14.8 months, respectively (NCT03575351). Tecartus: overall survival of 25.5 months and remission rate of 85% at 24 weeks [40]. | Commercially Available Some PI/IICTs: (NCT04227015) (NCT04844086) (NCT05470777) |
| BCMA-targeting CARs (Abecma, Carvykti) | Multiple myeloma | Abecma: overall and progression-free survival of 12.5 months and 8.5 months, respectively. A total of 82% of patients developed some grade of CRS, but only 3% had it at grade 3 or higher [41]. Carvykti: very good partial responses or complete responses were achieved in 66.7% of patients in a PIICT (NCT4133636). After 13.5 months, only one patient retained a clinical response. A total of 83.3% of the patients presented with some grade of CRS, up to grade 4 for one patient. | Commercially Available Some PI/IICTs: (NCT03767751) (NCT03448978) (NCT03361748) |
| CD20-targeting CARs | Various lymphomas and leukemias | Six complete remissions, three partial remissions and two stable diseases in 11 patients in a PIICT (NCT01735604), with a median progression-free survival of 6 months. | Some PI/IICTs: (NCT04007029) (NCT04697940) |
| CD22-targeting CARs | Various lymphomas and leukemias | In a PICT (NCT04088890), three patients (100%) with recurrent malignancies after CD19-targeting CAR T cell therapy achieved complete remission. Adverse events such as grade 1 and 2 CRS and high-grade neutropenia, thrombocytopenia, and anemia were detected. | Some PI/IICTs: (NCT05470777) (NCT05507827) |
| IL13Rα2-targeting CARs | Glioblastoma | A 228-day-long regression in one patient (PICT NCT02208362). Recurrence of cancer at four new locations, probably due to reduced TAA expression [42]. | Some PICTs: (NCT04003649) (NCT04661384) (NCT02208362) |
| Allogeneic NKG2D-based CAR (CYAD-101) | Metastatic colorectal cancer | In a PICT (NCT03692429) of 15 patients, 2 partial responses and 9 stable diseases were achieved, 7 of which lasted at least 3 months. Median progression-free survival: 3.9 months [43]. | Some PICTs: (NCT03692429) (NCT04991948) |
| HER2-targeting CARs | HER2+ cancers (pancreatic, breast, gastric, others) | In an advanced pancreatic cancer PICT (NCT01935843) of 11 patients, a 4.5 month-long partial response and 5 stable diseases were achieved. Median progression-free survival: 4.8 months (range, 1.5–8.3 months) [44]. | Some PI/IICTs: (NCT04650451) (NCT04430595) (NCT04650451) |
| GPC3-targeting CARs | Hepatocellular carcinoma | In two advanced hepatocellular carcinoma PICTs (NCT02395250 and NCT03146234), of a total of 13 patients, 2 partial responses were obtained. One patient with stable disease was alive after 44.2 months. Overall survival: 50.3% (6 months), 10.5% (3 years). One grade 5 and several grade 1/2 CRS events were recorded [45]. | Some PI/IICTs: (NCT03198546) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Marco, R.C.; Monzo, H.J.; Ojala, P.M. CAR T Cell Therapy: A Versatile Living Drug. Int. J. Mol. Sci. 2023, 24, 6300. https://doi.org/10.3390/ijms24076300
De Marco RC, Monzo HJ, Ojala PM. CAR T Cell Therapy: A Versatile Living Drug. International Journal of Molecular Sciences. 2023; 24(7):6300. https://doi.org/10.3390/ijms24076300
Chicago/Turabian StyleDe Marco, Rodrigo C., Hector J. Monzo, and Päivi M. Ojala. 2023. "CAR T Cell Therapy: A Versatile Living Drug" International Journal of Molecular Sciences 24, no. 7: 6300. https://doi.org/10.3390/ijms24076300