
Genome-Wide Association Study for Seed Dormancy Using Re-Sequenced Germplasm under Multiple Conditions in Rice

Abstract
:1. Introduction
2. Results
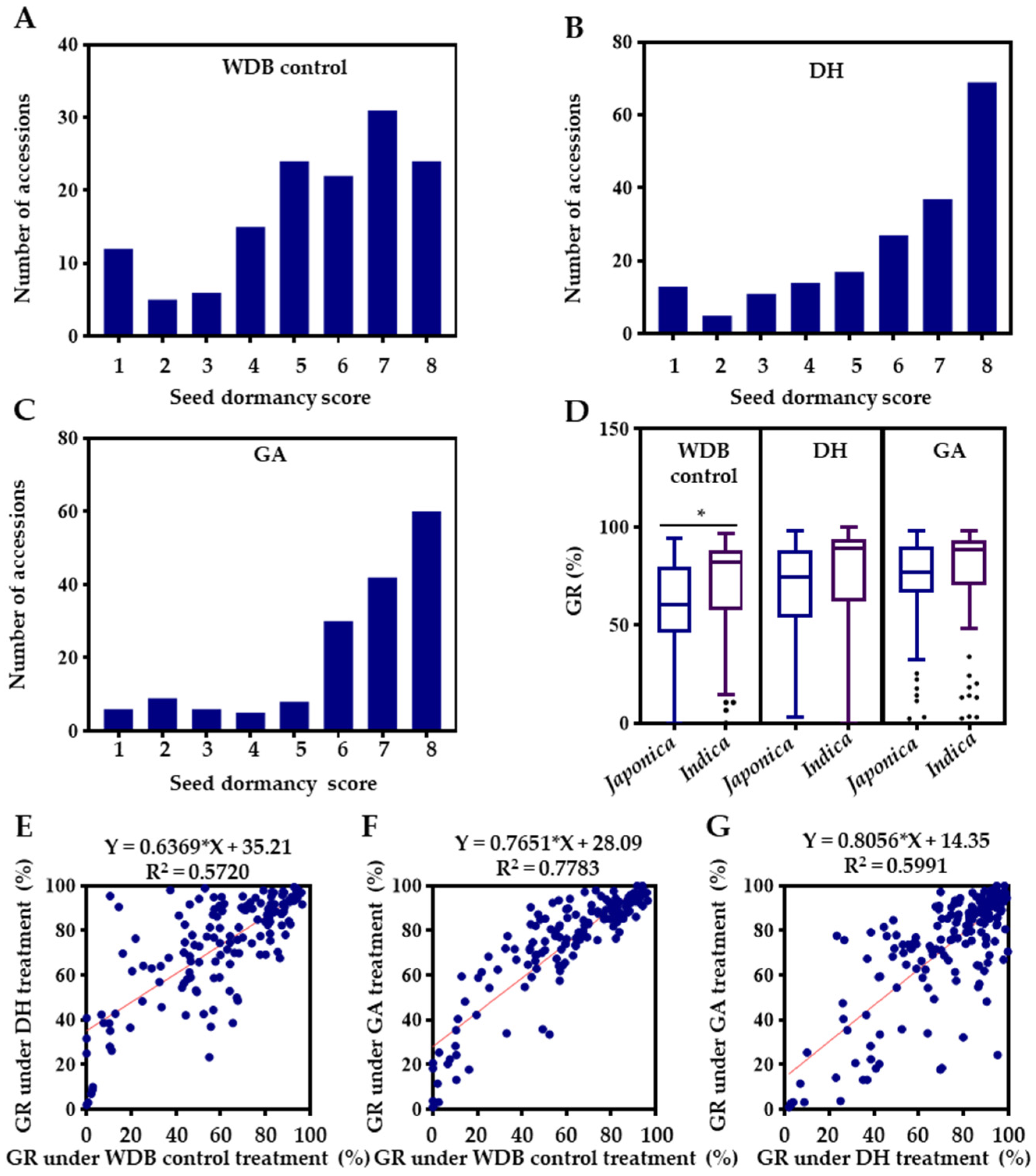
2.1. Phenotypic Analysis of Seed Dormancy
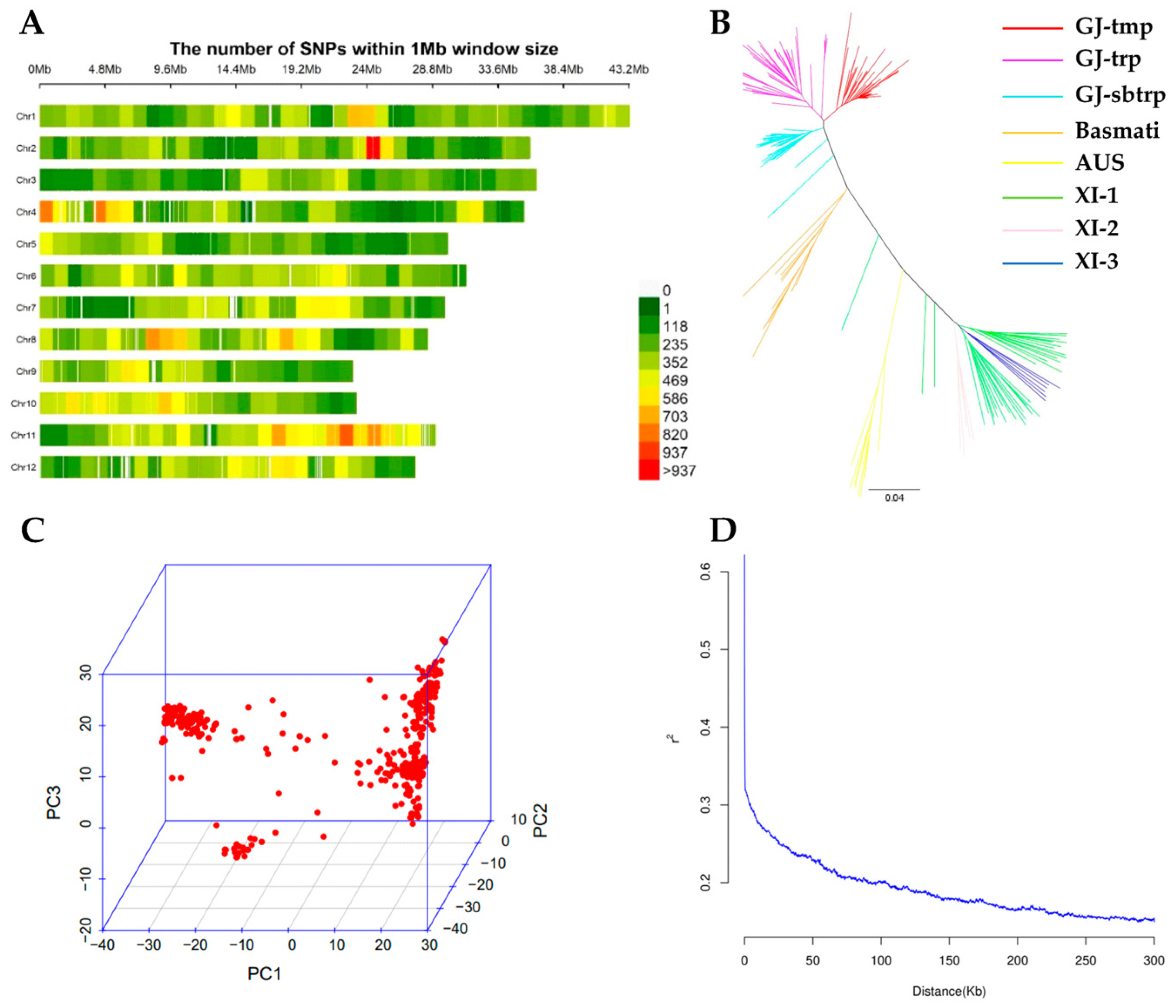
2.2. Single Nucleotide Polymorphism (SNP) Data, Population Structure and LD Pattern in the Panel
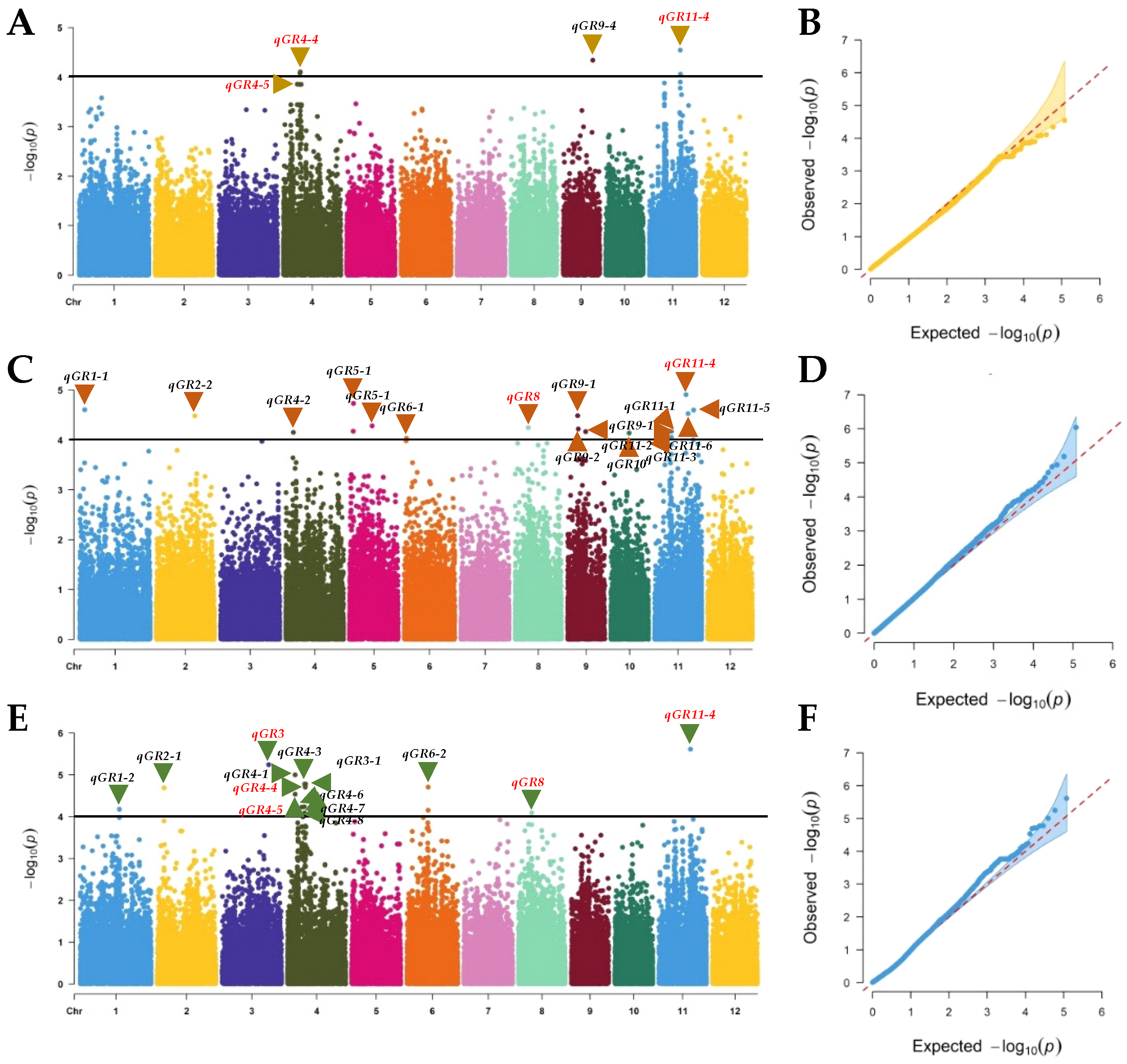
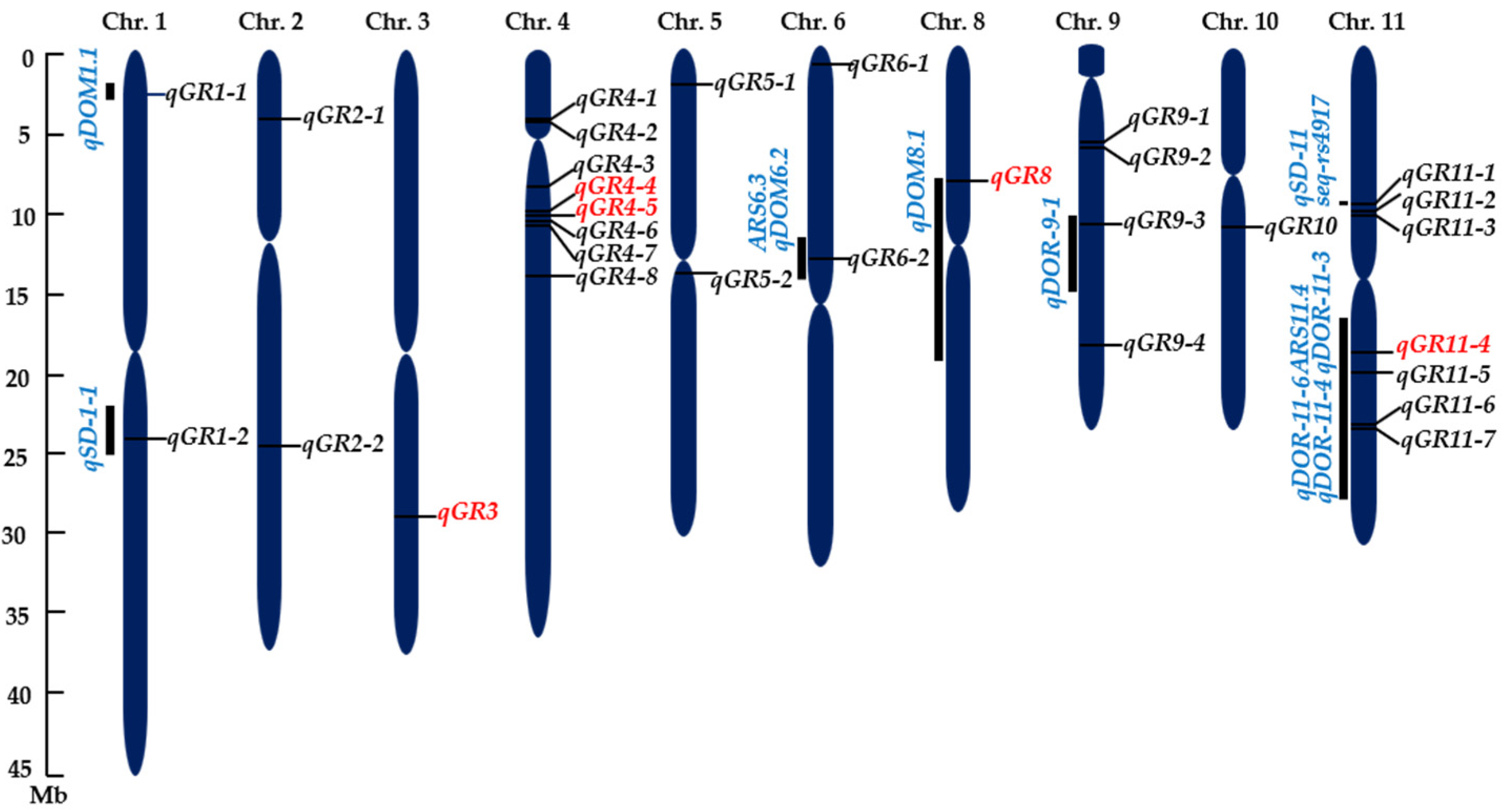
2.3. GWAS of Seed Dormancy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTL | Treatment | Chr. | Peak SNP | Effect | SE | p-Value | R2 | Germination Indicator Used in Previous Study | References |
|---|---|---|---|---|---|---|---|---|---|
| qGR1-1 | DH | 1 | 2,998,209 | 1.0 | 3.8 | 2.49 × 10−5 | 10.8% | T50 (time to reach 50% germination of the total number of germinated seeds). U8416 (time interval between 84% and 16% of viable seed to germinate). Intact seed harvested on 35 day after flowering. | [28] |
| qGR1-2 | GA | 1 | 23,870,065 | 18.4 | 5.3 | 6.68 × 10−5 | 16.8% | GR of intact seed (35 day after heading (DAH)) at day seven. | [41] |
| qGR2-1 | GA | 2 | 4,193,857 | 23.8 | 6.5 | 2.04 × 10−5 | 13.8% | - | - |
| qGR2-2 | DH | 2 | 23,728,701 | −15.5 | 5.9 | 3.31 × 10−5 | 9.3% | - | - |
| qGR3 | GA | 3 | 28,515,163 | −24.0 | 6.8 | 5.69 × 10−5 | 18.7% | - | - |
| qGR4-1 | GA | 4 | 4,523,405 | 19.3 | 5.5 | 1.00 × 10−5 | 17.8% | - | - |
| qGR4-2 | DH | 4 | 4,662,452 | 17.2 | 6.8 | 7.07 × 10−5 | 8.7% | - | - |
| qGR4-3 | GA | 4 | 8,563,180 | −18.3 | 6.4 | 5.97 × 10−5 | 14.8% | - | - |
| qGR4-4 | WDB control | 4 | 10,123,838 | 27.7 | 7.1 | 8.32 × 10−5 | 17.5% | - | - |
| GA | 4 | 10,123,838 | 27.7 | 7.1 | 5.79 × 10−5 | 14.8% | - | - | |
| qGR4-5 | GA | 4 | 10,352,980 | −24.0 | 9.1 | 1.66 × 10−5 | 16.9% | - | - |
| WDB control | 4 | 10,355,218 | 20.6 | 7.3 | 7.79 × 10−5 | 20.4% | - | - | |
| qGR4-6 | GA | 4 | 10,726,116 | −23.8 | 7.2 | 1.95 × 10−5 | 16.6% | - | - |
| qGR4-7 | GA | 4 | 10,981,781 | 24.0 | 9.1 | 1.66 × 10−5 | 16.9% | - | - |
| qGR4-8 | GA | 4 | 14,056,148 | 6.6 | 6.6 | 7.05 × 10−5 | 14.5% | - | - |
| qGR5-1 | DH | 5 | 2,322,980 | 1.0 | 5.1 | 1.85 × 10−5 | 9.8% | - | - |
| qGR5-2 | DH | 5 | 13,914,542 | 7.7 | 4.1 | 5.21 × 10−5 | 8.9% | - | - |
| qGR6-1 | DH | 6 | 1,484,749 | 3.8 | 3.6 | 9.29 × 10−5 | 9.6% | - | - |
| qGR6-2 | GA | 6 | 13,120,454 | 27.6 | 7.2 | 1.95 × 10−5 | 16.6% | GR at day seven. GR at day three. T50. U8416. AUC (area under the germination curve). GI (germination index). GR of after-ripened intact seed (32 DAH) at day seven. | [28,34] |
| qGR8 | DH | 8 | 8,040,564 | 24.5 | 11.6 | 5.68 × 10−5 | 8.9% | GR at day three. T50. | [28] |
| GA | 8 | 8,073,758 | −30.7 | 7.5 | 8.14 × 10−5 | 14.3% | |||
| qGR9-1 | DH | 9 | 6,327,905 | −29.9 | 9.5 | 3.27 × 10−5 | 10.5% | - | - |
| qGR9-2 | DH | 9 | 6,672,606 | 5.2 | 5.2 | 6.01 × 10−5 | 8.8% | - | - |
| qGR9-3 | DH | 9 | 11,261,867 | 10.2 | 10.6 | 6.80 × 10−5 | 8.7% | GR of intact seed and de-hulled seed (30 DAH) at day seven. | [42] |
| qGR9-4 | WDB control | 9 | 18,641,166 | −24.8 | 7.3 | 4.53 × 10−5 | 15.4% | - | - |
| qGR10 | DH | 10 | 11,242,705 | 0.7 | 4.5 | 7.28 × 10−5 | 8.6% | - | - |
| qGR11-1 | DH | 11 | 9,730,691 | −3.0 | 7.6 | 6.57 × 10−5 | 7.3% | GR of intact seed (30–40 DAH) at ten days. GR of intact seed (effective cumulative temperature reached 600° after heading) at day seven. | [2,29] |
| qGR11-2 | DH | 11 | 10,370,801 | −9.4 | 5.1 | 8.89 × 10−5 | 9.6% | - | - |
| qGR11-3 | DH | 11 | 10,544,254 | 10.2 | 5.6 | 6.75 × 10−5 | 9.9% | - | - |
| qGR11-4 | WDB control | 11 | 19,263,268 | −23.5 | 9.0 | 2.84 × 10−5 | 16.3% | GR of after-ripened intact seed (32 DAH) at day seven. GR of intact seed (30 DAH) at day seven. | [34,42] |
| DH | 11 | 19,313,112 | −6.4 | 6.6 | 1.23 × 10−5 | 10.1% | |||
| GA | 11 | 19,313,112 | −6.4 | 6.6 | 2.44 × 10−5 | 20.2% | |||
| qGR11-5 | DH | 11 | 20,619,141 | 19.9 | 9.1 | 2.97 × 10−5 | 9.4% | GR of de-hulled seed (60 DAH) at day seven. | [42] |
| qGR11-6 | DH | 11 | 23,872,614 | 4.6 | 4.4 | 9.96 × 10−5 | 9.5% | GR of intact seed (30 and 60 DAH) at day seven. | [42] |
| qGR11-7 | DH | 11 | 24,096,053 | −1.9 | 3.8 | 2.52 × 10−5 | 10.7% | GR of intact seed (30 and 60 DAH) at day seven. | [42] |
2.4. Candidate Gene Prediction
| QTL | Candidate Genes | Chr. | Functional Annotation | log2(12 h/0 h) * | log2(24 h/0 h) * | References |
|---|---|---|---|---|---|---|
| qGR3 | LOC_Os03g49990 | 3 | GRAS family transcription factor domain containing protein, expressed | 0.4 | NA | - |
| LOC_Os03g50050 | 3 | OsFBDUF16-F-box and DUF domain containing protein, expressed | −0.4 | −0.5 | [44,45,46,47] | |
| LOC_Os03g50110 | 3 | Similar to GEBP transcription factor (Fragment) | −1.9 | −1.9 | [48,49,50,51,52] | |
| LOC_Os03g50120 | 3 | Zinc finger family protein, putative, expressed | −0.4 | −0.9 | - | |
| LOC_Os03g50130 | 3 | Microsomal glutathione S-transferase 3, putative, expressed | NA | 1 | [53,54,55,56] | |
| LOC_Os03g50140 | 3 | Plastocyanin-like domain containing protein, putative, expressed | 4.3 | 4.2 | [57] | |
| LOC_Os03g50160 | 3 | Plastocyanin-like domain containing protein, putative, expressed | 4.5 | 3.7 | [57] | |
| qGR4-4 | LOC_Os04g18200 | 4 | Dihydrodipicolinate synthase, chloroplast precursor, putative, expressed | −1.2 | −1.2 | [58,59,60,61] |
| LOC_Os04g18380 | 4 | Cytochrome P450, putative, expressed | NA | NA | [62,63] | |
| qGR4-5 | LOC_Os04g18650 | 4 | AP2 domain containing protein, expressed | −0.4 | −0.3 | [64,65,66] |
| LOC_Os04g18790 | 4 | OsFBX126-F-box domain containing protein, expressed | NA | NA | [44,45,46,47] | |
| qGR8 | LOC_Os08g13360 | 8 | Kelch repeat protein, putative, expressed | −0.5 | −0.4 | - |
| LOC_Os08g13440 | 8 | Cupin domain containing protein, expressed | NA | 0.7 | - | |
| LOC_Os08g13640 | 8 | Ankyrin, putative, expressed | −0.9 | −1 | [67,68] | |
| qGR11-4 | LOC_Os11g32510 | 11 | OsGH3-13-Auxin-responsive GH3 gene family member, expressed | NA | 0.6 | - |
| LOC_Os11g32610 | 11 | Chalcone and stilbene synthases, putative, expressed | NA | NA | - | |
| LOC_Os11g32650 | 11 | Chalcone synthase, putative, expressed | 3.1 | NA | [69,70,71,72] | |
| LOC_Os11g32720 | 11 | Zinc knuckle domain containing protein, expressed | NA | −0.4 | - | |
| LOC_Os11g32750 | 11 | Hydrolase, NUDIX family, domain containing protein, expressed | 1.3 | 1.8 | [73,74] | |
| LOC_Os11g32810 | 11 | Leucine Rich Repeat family protein, expressed | −0.3 | −0.8 | [75,76,77] |
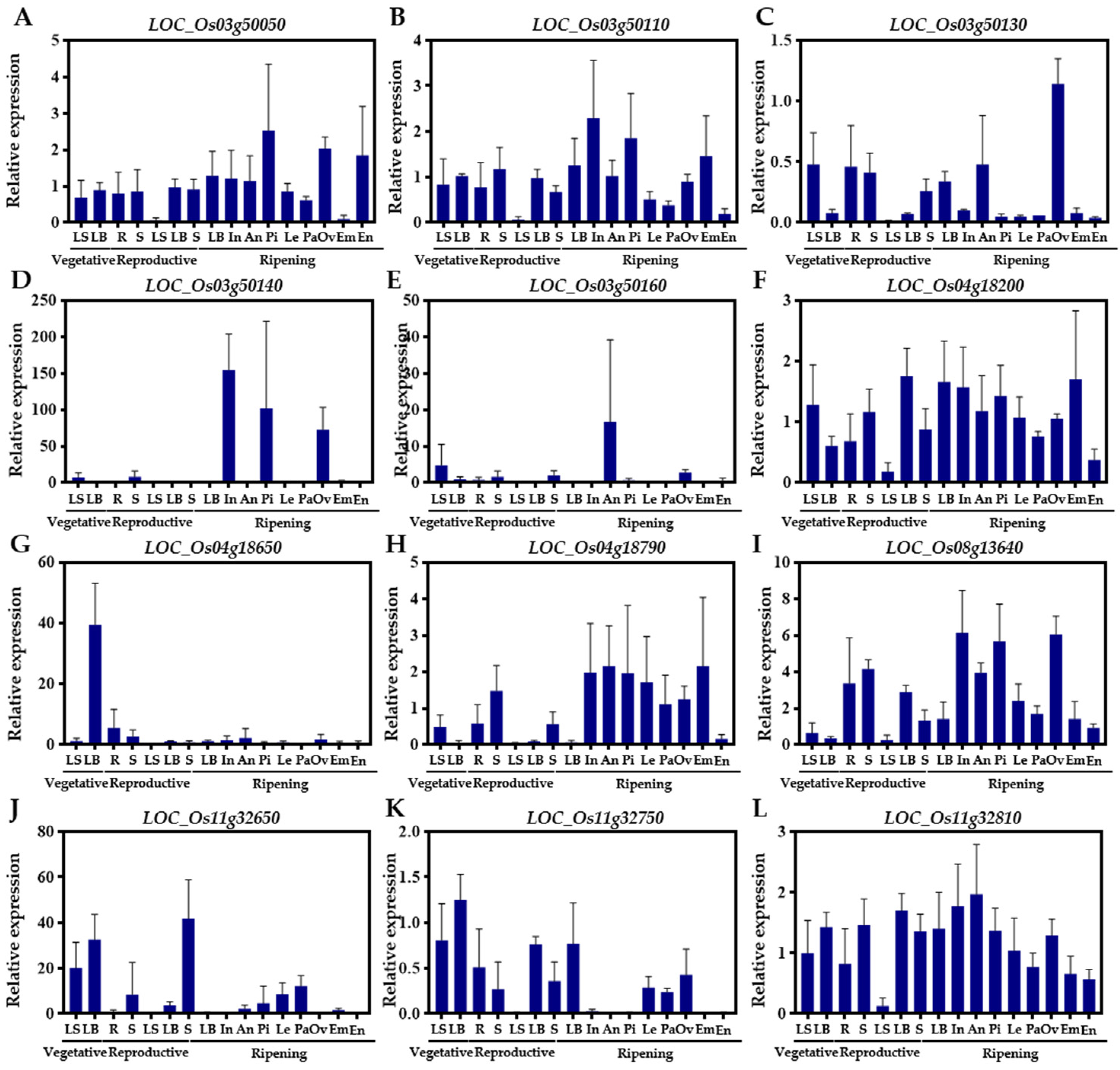
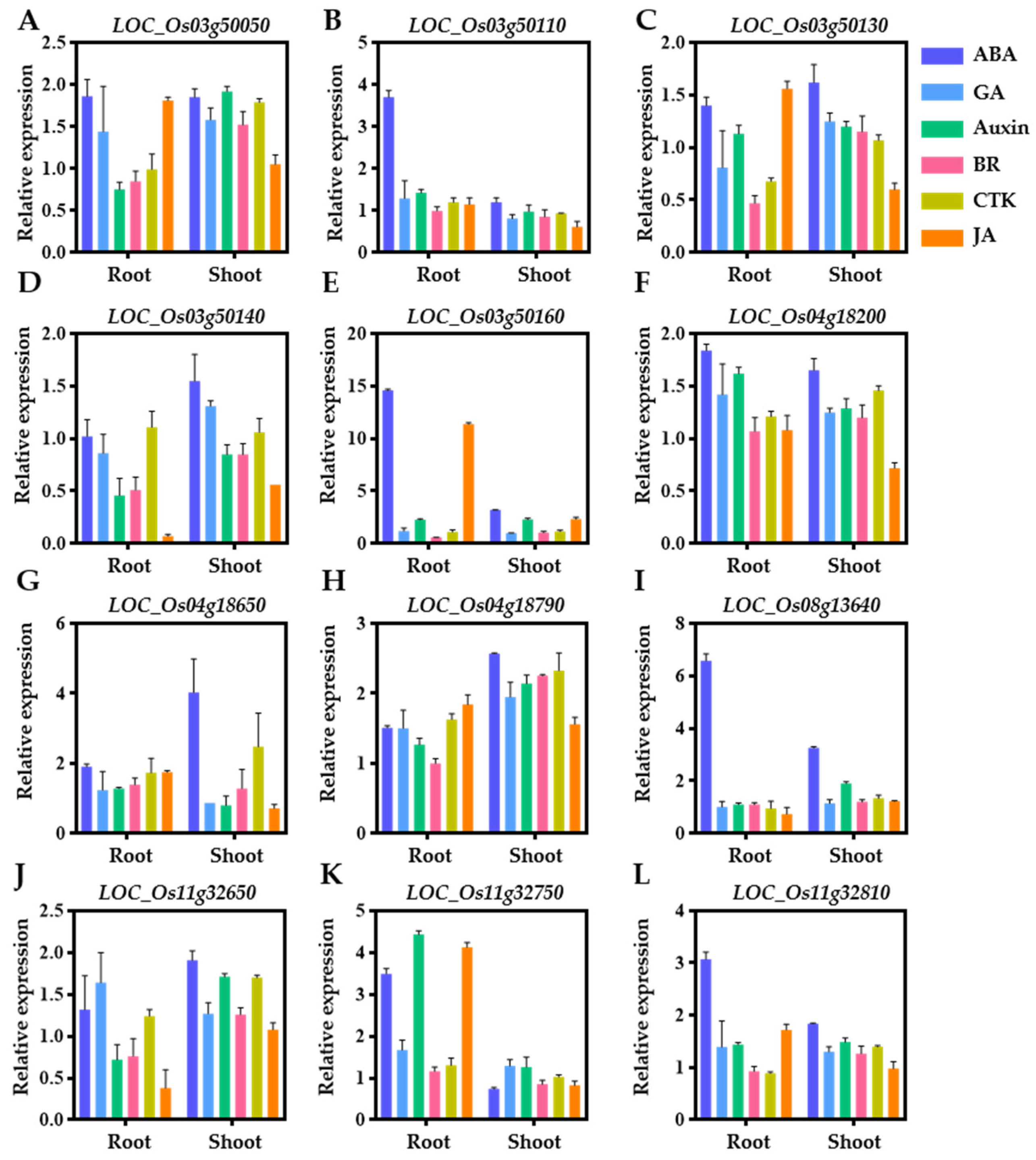
2.5. Expression Pattern throughout the Growth Period and Hormone Response of Candidate Genes
3. Discussion
3.1. Phenotypic Variation of Seed Dormancy
3.2. GWAS of Rice Seed Dormancy
3.3. Candidate Genes for Seed Dormancy
4. Materials and Methods
4.1. Plant Material and Growth Conditions for Multiplication
4.2. Assessment of Seed Germination
4.3. Genotyping
4.4. Population Structure Analysis
4.5. Linkage Disequilibrium Analysis
4.6. Genome-Wide Association Analysis
4.7. Candidate Gene Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shu, K.; Liu, X.D.; Xie, Q.; He, Z.H. Two Faces of One Seed: Hormonal Regulation of Dormancy and Germination. Mol. Plant 2016, 9, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Niu, X.J.; Zhang, M.C.; Wang, C.H.; Xu, Q.; Feng, Y.; Yang, Y.L.; Wang, S.; Yuan, X.P.; Yu, H.Y.; et al. Genome-Wide Association Study of Seed Dormancy and the Genomic Consequences of Improvement Footprints in Rice (Oryza sativa L.). Front. Plant Sci. 2018, 8, 2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.Y.; Foley, M.E.; Horvath, D.P.; Anderson, J.V.; Feng, J.H.; Zhang, L.H.; Mowry, C.R.; Ye, H.; Suttle, J.C.; Kadowaki, K.; et al. Association Between Seed Dormancy and Pericarp Color Is Controlled by a Pleiotropic Gene That Regulates Abscisic Acid and Flavonoid Synthesis in Weedy Red Rice. Genetics 2011, 189, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Baskin, C.C.; Baskin, J.M. Introduction. Seeds: Ecology, Biogeography, and Evolution of Dormancy and Germination, 2nd ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Gupta, S.; Doležal, K.; Kulkarni, M.G.; Balázs, E.; Van Staden, J. Role of non-microbial biostimulants in regulation of seed germination and seedling establishment. Plant Growth Regul. 2022, 97, 271–313. [Google Scholar] [CrossRef]
- Gubler, F.; Millar, A.A.; Jacobsen, J.V. Dormancy release, ABA and pre-harvest sprouting. Curr. Opin. Plant Biol. 2005, 8, 183–187. [Google Scholar] [CrossRef]
- Zhu, D.W.; Qian, Z.H.; Wei, H.Y.; Guo, B.W.; Xu, K.; Dai, Q.G.; Zhang, H.C.; Huo, Z.Y. The effects of field pre-harvest sprouting on the morphological structure and physicochemical properties of rice (Oryza sativa L.) starch. Food Chem. 2019, 278, 10–16. [Google Scholar] [CrossRef]
- Tai, L.; Wang, H.J.; Xu, X.J.; Sun, W.H.; Ju, L.; Liu, W.T.; Li, W.Q.; Sun, J.Q.; Chen, K.M. Pre-harvest sprouting in cereals: Genetic and biochemical mechanisms. J. Exp. Bot. 2021, 72, 2857–2876. [Google Scholar] [CrossRef]
- Black, M.; Bewley, J.D.; Halmer, P. Encyclopedia of Seeds—Science, Technology and Uses; CABI: Wallingford, UK, 2006. [Google Scholar]
- Muthayya, S.; Sugimoto, J.D.; Montgomery, S.; Maberly, G.F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 2014, 1324, 7–14. [Google Scholar] [CrossRef]
- Guo, L.B.; Ye, G.Y. Use of Major Quantitative Trait Loci to Improve Grain Yield of Rice. Rice Sci. 2014, 21, 65–82. [Google Scholar] [CrossRef]
- Gianinetti, A.; Vernieri, P. On the role of abscisic acid in seed dormancy of red rice. J. Exp. Bot. 2007, 58, 3449–3462. [Google Scholar] [CrossRef] [Green Version]
- Yan, A.; Chen, Z. The Control of Seed Dormancy and Germination by Temperature, Light and Nitrate. Bot. Rev. 2020, 86, 39–75. [Google Scholar] [CrossRef]
- Barrero, J.M.; Downie, A.B.; Xu, Q.; Gubler, F. A Role for Barley CRYPTOCHROME1 in Light Regulation of Grain Dormancy and Germination. Plant Cell 2014, 26, 1094–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, B. Genomic architecture of promoters and transcriptional regulation of candidate genes in rice involved in tolerance to anaerobic germination. Curr. Plant Biol. 2022, 29, 100236. [Google Scholar] [CrossRef]
- Seshu, D.V.; Dadlani, M. Mechanism of seed dormancy in rice. Seed Sci. Res. 1991, 1, 187–194. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, Q.B.; Wu, T.; Duan, E.C.; Huang, Y.S.; Yang, C.Y.; Mou, C.L.; Lan, J.; Zhou, C.L.; Xie, K.; et al. OsDOG1L-3 regulates seed dormancy through the abscisic acid pathway in rice. Plant Sci. 2020, 298, 110570. [Google Scholar] [CrossRef]
- Sohn, S.I.; Pandian, S.; Kumar, T.S.; Zoclanclounon, Y.A.B.; Muthuramalingam, P.; Shilpha, J.; Satish, L.; Ramesh, M. Seed Dormancy and Pre-Harvest Sprouting in Rice-An Updated Overview. Int. J. Mol. Sci. 2021, 22, 11804. [Google Scholar] [CrossRef]
- Wan, J.; Nakazaki, T.; Kawaura, K.; Ikehashi, H. Identification of marker loci for seed dormancy in rice (Oryza sativa L.). Crop Sci. 1997, 37, 1759–1763. [Google Scholar] [CrossRef]
- Lu, B.Y.; Xie, K.; Yang, C.Y.; Wang, S.F.; Liu, X.; Zhang, L.; Jiang, L.; Wan, J.M. Mapping two major effect grain dormancy QTL in rice. Mol. Breed. 2011, 28, 453–462. [Google Scholar] [CrossRef]
- Graeber, K.; Nakabayashi, K.; Miatton, E.; Leubner-Metzger, G.; Soppe, W.J.J. Molecular mechanisms of seed dormancy. Plant Cell Environ. 2012, 35, 1769–1786. [Google Scholar] [CrossRef]
- Xu, F.; Tang, J.Y.; Wang, S.X.; Cheng, X.; Wang, H.R.; Ou, S.J.; Gao, S.P.; Li, B.S.; Qian, Y.W.; Gao, C.X.; et al. Antagonistic control of seed dormancy in rice by two bHLH transcription factors. Nat. Genet. 2022, 54, 1972–1982. [Google Scholar] [CrossRef]
- Phan, P.D.T.; Vu, B.V. Improving the pre-harvest sprouting resistance of rice cultivar IR36 using wild rice (Oryza rufipogon) W630. Cereal Res. Commun. 2022, 50, 37–43. [Google Scholar] [CrossRef]
- Cheon, K.S.; Won, Y.J.; Jeong, Y.M.; Lee, Y.Y.; Kang, D.Y.; Oh, J.; Oh, H.; Kim, S.L.; Kim, N.; Lee, E.; et al. QTL mapping for pre-harvest sprouting resistance in japonica rice varieties utilizing genome re-sequencing. Mol. Genet. Genom. 2020, 295, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Wang, H.; Zhao, C.Z.; Tonnis, B.; Tallury, S.; Wang, X.J.; Clevenger, J.; Guo, B.Z. Identification of QTLs for Seed Dormancy in Cultivated Peanut Using a Recombinant Inbred Line Mapping Population. Plant Mol. Biol. Rep. 2022, 40, 208–217. [Google Scholar] [CrossRef]
- Hori, K.; Sugimoto, K.; Nonoue, Y.; Ono, N.; Matsubara, K.; Yamanouchi, U.; Abe, A.; Takeuchi, Y.; Yano, M. Detection of quantitative trait loci controlling pre-harvest sprouting resistance by using backcrossed populations of japonica rice cultivars. Theor. Appl. Genet. 2010, 120, 1547–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.L.; Xie, Y.H.; Cai, J.L.; Liu, C.B.; Zhu, H.T.; Jiang, R.; Zhong, Y.Y.; Zhang, G.L.; Tan, B.; Liu, G.F.; et al. Substitution mapping of QTLs controlling seed dormancy using single segment substitution lines derived from multiple cultivated rice donors in seven cropping seasons. Theor. Appl. Genet. 2017, 130, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.W.; Wang, Y.T.; Zhang, C.P.; He, H.Z.; Yu, S.B. Genetic Dissection of Seed Dormancy using Chromosome Segment Substitution Lines in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2020, 21, 1344. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.B.; Zhu, L.H.; Xu, Y.B.; Zeng, D.L.; Wu, P.; Qian, Q. QTL analysis of seed dormancy in rice (Oryza sativa L.). Euphytica 2004, 140, 155–162. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Lin, S.Y.; Sasaki, T.; Yano, M. Fine linkage mapping enables dissection of closely linked quantitative trait loci for seed dormancy and heading in rice. Theor. Appl. Genet. 2003, 107, 1174–1180. [Google Scholar] [CrossRef]
- Gu, X.Y.; Liu, T.L.; Feng, J.H.; Suttle, J.C.; Gibbons, J. The qSD12 underlying gene promotes abscisic acid accumulation in early developing seeds to induce primary dormancy in rice. Plant Mol. Biol. 2010, 73, 97–104. [Google Scholar] [CrossRef]
- Ye, H.; Foley, M.E.; Gu, X.Y. New seed dormancy loci detected from weedy rice-derived advanced populations with major QTL alleles removed from the background. Plant Sci. 2010, 179, 612–619. [Google Scholar] [CrossRef]
- Nguyen, T.; Fu, K.; Mou, C.; Yu, J.; Zhu, X.; Huang, Y.; Zhou, C.; Hao, Q.; Zhang, F.; Song, W.; et al. Fine mapping of qSdr9, a novel locus for seed dormancy (SD) in weedy rice, and development of NILs with a strong SD allele. Mol. Breed. 2020, 40, 81. [Google Scholar] [CrossRef]
- Magwa, R.A.; Zhao, H.; Xing, Y.Z. Genome-wide association mapping revealed a diverse genetic basis of seed dormancy across subpopulations in rice (Oryza sativa L.). BMC Genet. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Shi, J.; Liang, W.; Zhang, D. Integrating GWAS and transcriptomics to identify genes involved in seed dormancy in rice. Appl. Genet. 2021, 134, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Takeuchi, Y.; Ebana, K.; Miyao, A.; Hirochika, H.; Hara, N.; Ishiyama, K.; Kobayashi, M.; Ban, Y.; Hattori, T.; et al. Molecular cloning of Sdr4, a regulator involved in seed dormancy and domestication of rice. Proc. Natl. Acad. Sci. USA 2010, 107, 5792–5797. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Deng, Q.W.; Li, Y.H.; Yu, Y.; Liu, X.; Han, Y.F.; Luo, X.D.; Wu, X.J.; Ju, L.; Sun, J.Q.; et al. Transcription Factors Rc and OsVP1 Coordinately Regulate Preharvest Sprouting Tolerance in Red Pericarp Rice. J. Agric. Food Chem. 2020, 68, 14748–14757. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Feng, J.H.; Zhang, L.H.; Zhang, J.F.; Mispan, M.S.; Cao, Z.Q.; Beighley, D.H.; Yang, J.C.; Gu, X.Y. Map-Based Cloning of Seed Dormancy1-2 Identified a Gibberellin Synthesis Gene Regulating the Development of Endosperm-Imposed Dormancy in Rice. Plant Physiol. 2015, 169, 2152–2165. [Google Scholar]
- Wang, L.; Cheng, J.P.; Lai, Y.Y.; Du, W.L.; Huang, X.; Wang, Z.F.; Zhang, H.S. Identification of QTLs with additive, epistatic and QTL x development interaction effects for seed dormancy in rice. Planta 2014, 239, 411–420. [Google Scholar] [CrossRef]
- Sweeney, M.; Mccouch, S. The complex history of the domestication of rice. Ann. Bot. 2007, 100, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Cao, Y.J.; Wang, C.M.; Zhai, H.Q.; Wan, J.M.; Yoshimura, A. Detection and analysis of QTL for seed dormancy in rice (Oryza sativa L.) using RIL and CSSL population. Yi Chuan Xue Bao 2003, 30, 453–458. [Google Scholar]
- Cai, H.W.; Morishima, H. Genomic regions affecting seed shattering and seed dormancy in rice. Theor. Appl. Genet. 2000, 100, 840–846. [Google Scholar] [CrossRef]
- Huang, S.; Taylor, N.L.; Narsai, R.; Eubel, H.; Whelan, J.; Millar, A.H. Experimental analysis of the rice mitochondrial proteome, its biogenesis, and heterogeneity. Plant Physiol. 2009, 149, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariizumi, T.; Lawrence, P.K.; Steber, C.M. The Role of Two F-Box Proteins, SLEEPY1 and SNEEZY, in Arabidopsis Gibberellin Signaling. Plant Physiol. 2011, 155, 765–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.Y.; Dai, X.Y.; Zhang, W.H. A rice F-box gene, OsFbx352, is involved in glucose-delayed seed germination in rice. J. Exp. Bot. 2012, 63, 5559–5568. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Sun, M.S.; Li, X.M.; He, R.Q.; Zhong, M.; Luo, D.; Liu, X.M.; Zhao, X.Y. The Arabidopsis F-box protein FOF2 regulates ABA-mediated seed germination and drought tolerance. Plant Sci. 2020, 301, 110643. [Google Scholar] [CrossRef]
- Kim, H.; Song, E.; Kim, Y.; Choi, E.; Hwang, J.; Lee, J.H. Loss-of-function of Arabidopsis F-box protein hypersensitive to ABA 1 enhances drought tolerance and delays germination. Physiol. Plant. 2021, 173, 2376–2389. [Google Scholar] [CrossRef]
- Riefler, M.; Novak, O.; Strnad, M.; Schmulling, T. Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 2006, 18, 40–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, F.; Perazza, D.; Laporte, F.; Le Henaff, G.; Hornitschek, P.; Bonneville, J.M.; Herzog, M.; Vachon, G. GeBP and GeBP-like proteins are noncanonical leucine-zipper transcription factors that regulate cytokinin response in Arabidopsis. Plant Physiol. 2008, 146, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Argueso, C.T.; Raines, T.; Kieber, J.J. Cytokinin signaling and transcriptional networks. Curr. Opin. Plant Biol. 2010, 13, 533–539. [Google Scholar] [CrossRef]
- Wang, Y.P.; Li, L.; Ye, T.T.; Zhao, S.J.; Liu, Z.; Feng, Y.Q.; Wu, Y. Cytokinin antagonizes ABA suppression to seed germination of Arabidopsis by downregulating ABI5 expression. Plant J. 2011, 68, 249–261. [Google Scholar] [CrossRef]
- Guan, C.M.; Wang, X.C.; Feng, J.; Hong, S.L.; Liang, Y.; Ren, B.; Zuo, J.R. Cytokinin Antagonizes Abscisic Acid-Mediated Inhibition of Cotyledon Greening by Promoting the Degradation of ABSCISIC ACID INSENSITIVE5 Protein in Arabidopsis. Plant Physiol. 2014, 164, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Jiang, H.W.; Hsieh, E.J.; Chen, H.Y.; Chien, C.T.; Hsieh, H.L.; Lin, T.P. Drought and Salt Stress Tolerance of an Arabidopsis Glutathione S-Transferase U17 Knockout Mutant Are Attributed to the Combined Effect of Glutathione and Abscisic Acid. Plant Physiol. 2012, 158, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Sahoo, A.; Devendran, R.; Jain, M. Over-Expression of a Rice Tau Class Glutathione S-Transferase Gene Improves Tolerance to Salinity and Oxidative Stresses in Arabidopsis. PLoS ONE 2014, 9, e92900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Tian, Y.S.; Xing, X.J.; Peng, R.H.; Zhu, B.; Gao, J.J.; Yao, Q.H. Over-expression of AtGSTU19 provides tolerance to salt, drought and methyl viologen stresses in Arabidopsis. Physiol. Plant. 2016, 156, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, N.; Liu, Z.G.; Liu, S.Y.; Liu, C.X.; Lin, J.H.; Yang, H.; Li, S.; Yukawa, Y. The AtGSTU7 gene influences glutathione-dependent seed germination under ABA and osmotic stress in Arabidopsis. Biochem. Biophys. Res. Commun. 2020, 528, 538–544. [Google Scholar] [CrossRef]
- Jiang, A.L.; Guo, Z.L.; Pan, J.W.; Yang, Y.Z.; Zhuang, Y.; Zuo, D.Q.; Hao, C.; Gao, Z.X.; Xin, P.Y.; Chu, J.F.; et al. The PIF1-miR408-PLANTACYANIN repression cascade regulates light-dependent seed germination. Plant Cell 2021, 33, 1506–1529. [Google Scholar] [CrossRef]
- Falco, S.C.; Guida, T.; Locke, M.; Mauvais, J.; Sanders, C.; Ward, R.T.; Webber, P. Transgenic Canola and Soybean Seeds with Increased Lysine. Biotechnology 1995, 13, 577–582. [Google Scholar] [CrossRef]
- Angelovici, R.; Fait, A.; Fernie, A.R.; Galili, G. A seed high-lysine trait is negatively associated with the TCA cycle and slows down Arabidopsis seed germination. New Phytol. 2011, 189, 148–159. [Google Scholar] [CrossRef]
- Qi, Q.G.; Huang, J.T.; Crowley, J.; Ruschke, L.; Goldman, B.S.; Wen, L.; Rapp, W.D. Metabolically engineered soybean seed with enhanced threonine levels: Biochemical characterization and seed-specific expression of lysine-insensitive variants of aspartate kinases from the enteric bacterium Xenorhabdus bovienii. Plant Biotechnol. J. 2011, 9, 193–204. [Google Scholar] [CrossRef]
- Atkinson, S.C.; Dogovski, C.; Downton, M.T.; Czabotar, P.E.; Dobson, R.C.J.; Gerrard, J.A.; Wagner, J.; Perugini, M.A. Structural, kinetic and computational investigation of Vitis vinifera DHDPS reveals new insight into the mechanism of lysine-mediated allosteric inhibition. Plant Mol. Biol. 2013, 81, 431–446. [Google Scholar] [CrossRef]
- Cutler, A.J.; Krochko, J.E. Formation and breakdown of ABA. Trends Plant Sci. 1999, 4, 472–478. [Google Scholar] [CrossRef]
- Saito, S.; Hirai, N.; Matsumoto, C.; Ohigashi, H.; Ohta, D.; Sakata, K.; Mizutani, M. Arabidopsis CYP707As encode (+)-abscisic acid 8′-hydroxylase, a key enzyme in the oxidative catabolism of abscisic acid. Plant Physiol. 2004, 134, 1439–1449. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, K.; Tatematsu, K.; Yano, R.; Preston, J.; Kitamura, S.; Takahashi, H.; McCourt, P.; Kamiya, Y.; Nambara, E. CHOTTO1, a Double AP2 Domain Protein of Arabidopsis thaliana, Regulates Germination and Seedling Growth under Excess Supply of Glucose and Nitrate. Plant Cell Physiol. 2009, 50, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Yano, R.; Kanno, Y.; Jikumaru, Y.; Nakabayashi, K.; Kamiya, Y.; Nambara, E. CHOTTO1, a Putative Double APETALA2 Repeat Transcription Factor, Is Involved in Abscisic Acid-Mediated Repression of Gibberellin Biosynthesis during Seed Germination in Arabidopsis. Plant Physiol. 2009, 151, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Yaish, M.W.; El-kereamy, A.; Zhu, T.; Beatty, P.H.; Good, A.G.; Bi, Y.M.; Rothstein, S.J. The APETALA-2-Like Transcription Factor OsAP2-39 Controls Key Interactions between Abscisic Acid and Gibberellin in Rice. PLoS Genet. 2010, 6, e1001098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, H.; Matsuda, O.; Iba, K. ITN1, a novel gene encoding an ankyrin-repeat protein that affects the ABA-mediated production of reactive oxygen species and is involved in salt-stress tolerance in Arabidopsis thaliana. Plant J. 2008, 56, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.Y.; Lu, Z.W.; Sun, Y.; Fang, Z.W.; Chen, J.; Zhou, Y.B.; Chen, M.; Ma, Y.Z.; Xu, Z.S.; Min, D.H. The Ankyrin-Repeat Gene GmANK114 Confers Drought and Salt Tolerance in Arabidopsis and Soybean. Front. Plant Sci. 2020, 11, 584167. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Delseny, M.; Devic, M. BANYULS, a novel negative regulator of flavonoid biosynthesis in the Arabidopsis seed coat. Plant J. 1997, 11, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Cheynier, V.; Comte, G.; Davies, K.M.; Lattanzio, V.; Martens, S. Plant phenolics: Recent advances on their biosynthesis, genetics, and ecophysiology. Plant Physiol. Biochem. 2013, 72, 1–20. [Google Scholar] [CrossRef]
- Chen, L.J.; Guo, H.M.; Lin, Y.; Cheng, H.M. Chalcone synthase EaCHS1 from Eupatorium adenophorum functions in salt stress tolerance in tobacco. Plant Cell Rep. 2015, 34, 885–894. [Google Scholar]
- Shen, Y.X.; Sun, T.T.; Pan, Q.; Anupol, N.; Chen, H.; Shi, J.W.; Liu, F.; Deqiang, D.M.; Wang, C.Q.; Zhao, J.; et al. RrMYB5-and RrMYB10-regulated flavonoid biosynthesis plays a pivotal role in feedback loop responding to wounding and oxidation in Rosa rugosa. Plant Biotechnol. J. 2019, 17, 2078–2095. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Li, Y.F.; Mahalingam, R. Arabidopsis nudix hydrolase 7 plays a role in seed germination. Planta 2014, 239, 1015–1025. [Google Scholar] [CrossRef]
- He, H.J.; Zhang, Y.Z.; Wen, B.B.; Meng, X.G.; Wang, N.; Sun, M.Y.; Zhang, R.; Zhao, X.H.; Tan, Q.P.; Xiao, W.; et al. PpNUDX8, a Peach NUDIX Hydrolase, Plays a Negative Regulator in Response to Drought Stress. Front. Plant Sci. 2022, 12, 831883. [Google Scholar] [CrossRef]
- Pitorre, D.; Llauro, C.; Jobet, E.; Guilleminot, J.; Brizard, J.P.; Delseny, M.; Lasserre, E. RLK7, a leucine-rich repeat receptor-like kinase, is required for proper germination speed and tolerance to oxidative stress in Arabidopsis thaliana. Planta 2010, 232, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gao, Z.R.; Xiao, G.Q.; Huang, R.F.; Zhang, H.W. Leucine-Rich Repeat Receptor-Like Kinase FON1 Regulates Drought Stress and Seed Germination by Activating the Expression of ABA-Responsive Genes in Rice. Plant Mol. Biol. Rep. 2014, 32, 1158–1168. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.C.; Yao, X.H.; Liu, S.H.; Zhang, P.Y.; Chen, K.S. The Antarctic moss leucine-rich repeat receptor-like kinase (PnLRR-RLK2) functions in salinity and drought stress adaptation. Polar Biol. 2018, 41, 353–364. [Google Scholar] [CrossRef]
- Yutaka, S.; Baltazar, A.; Nobukazu, N.; Ritsuko, M.; Kazuhiko, S.; Hinako, T.; Hiroshi, M.; Kaori, K.; Makoto, K.; Hirohiko, H.; et al. Field transcriptome revealed critical developmental and physiological transitions involved in the expression of growth potential in japonica rice. BMC Plant Biol. 2011, 11, 10. [Google Scholar]
- Tuan, P.A.; Kumar, R.; Rehal, P.K.; Toora, P.K.; Ayele, B.T. Molecular Mechanisms Underlying Abscisic Acid/Gibberellin Balance in the Control of Seed Dormancy and Germination in Cereals. Front. Plant Sci. 2018, 9, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.L.; Foley, M.E. Genetic and molecular control of seed dormancy. Trends Plant Sci. 1997, 2, 384–389. [Google Scholar] [CrossRef]
- Yang, J.; Yang, M.; Su, L.; Zhou, D.H.; Huang, C.H.; Wang, H.; Guo, T.; Chen, Z.Q. Genome-wide association study reveals novel genetic loci contributing to cold tolerance at the germination stage in indica rice. Plant Sci. 2020, 301, 110669. [Google Scholar] [CrossRef]
- Yang, B.; Chen, M.M.; Zhan, C.F.; Liu, K.X.; Cheng, Y.H.; Xie, T.; Zhu, P.W.; He, Y.; Zeng, P.; Tang, H.J.; et al. Identification of OsPK5 involved in rice glycolytic metabolism and GA/ABA balance for improving seed germination via genome-wide association study. J. Exp. Bot. 2022, 73, 3446–3461. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, Y.H.; Lee, G.S. Mapping QTLs associated to germination stability following dry-heat treatment in rice seed. 3 Biotech 2017, 7, 220. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yang, C.; Ding, B.; Feng, Z.; Wang, Q.; He, J.; Tong, J.; Xiao, L.; Jiang, L.; Wan, J. Microarray-based gene expression analysis of strong seed dormancy in rice cv. N22 and less dormant mutant derivatives. Plant Physiol. Biochem. 2016, 99, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Yang, J.; Li, D.X.; Sun, K.; Luo, L.X.; Xiao, W.M.; Wang, J.F.; Liu, Y.Z.; Wang, S.; Wang, H.; et al. Integrating GWAS, QTL, mapping and RNA-seq to identify candidate genes for seed vigor in rice (Oryza sativa L.). Mol. Breed. 2019, 39, 87. [Google Scholar] [CrossRef]
- Gupta, R.; Chakrabarty, S.K. Gibberellic acid in plant Still a mystery unresolved. Plant Signal. Behav. 2013, 8, e25504. [Google Scholar] [CrossRef] [Green Version]
- Sukifto, R.; Nulit, R.; Kong, Y.C.; Sidek, N.; Mahadi, S.N.; Mustafa, N.; Razak, R.A. Enhancing germination and early seedling growth of Malaysian indica rice (Oryza sativa L.) using hormonal priming with gibberellic acid (GA3). AIMS Agric. Food 2020, 5, 649–665. [Google Scholar] [CrossRef]
- Xu, Y.; Li, P.C.; Yang, Z.F.; Xu, C.W. Genetic mapping of quantitative trait loci in crops. Crop J. 2017, 5, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Naeem, M.; Demirel, U.; Yousaf, M.F.; Caliskan, S.; Caliskan, M.E. Overview on domestication, breeding, genetic gain and improvement of tuber quality traits of potato using fast forwarding technique (GWAS): A review. Plant Breed. 2021, 140, 519–542. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.S.; Mauleon, R.; Hu, Z.Q.; Chebotarov, D.; Tai, S.S.; Wu, Z.C.; Li, M.; Zheng, T.Q.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Cheng, J.; Wang, J.; Cheng, Y.; He, Y.; Zhang, H.; Wang, Z. Physiological characteristics of cold stratification on seed dormancy release in rice. Plant Growth Regul. 2019, 89, 131–141. [Google Scholar] [CrossRef]
- Gu, X.Y.; Kianian, S.F.; Foley, M.E. Multiple loci and epistases control genetic variation for seed dormancy in weedy rice (Oryza sativa). Genetics 2004, 166, 1503–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.P.; Wang, L.; Du, W.L.; Lai, Y.Y.; Huang, X.; Wang, Z.F.; Zhang, H.S. Dynamic quantitative trait locus analysis of seed dormancy at three development stages in rice. Mol. Breed. 2014, 34, 501–510. [Google Scholar] [CrossRef]
- Li, A.H.; Jiang, S.Y.; Yang, G.; Li, Y.; Guo, N.; Chen, T.; Kang, L.P.; Huang, L.Q. Molecular mechanism of seed dormancy release induced by fluridone compared with cod stratification in Notopterygium incisum. BMC Plant Biol. 2018, 18, 116. [Google Scholar]
- Dong, Y.J.; Tsuzuki, E.; Kamiunten, H.; Terao, H.; Lin, D.Z.; Matsuo, M.; Zheng, Y.F. Identification of quantitative trait loci associated with pre-harvest sprouting resistance in rice (Oryza sativa L.). Field Crops Res. 2003, 81, 133–139. [Google Scholar] [CrossRef]
- Khan, I.A.; Awan, F.S.; Ahmad, A.; Khan, A.A. A modified mini-prep method for economical and rapid extraction of genomic DNA in plants. Plant Mol. Biol. Rep. 2004, 22, 89. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| Trait | Seed Germination (%) | |||
|---|---|---|---|---|
| Treatment | Terms | Japonica | Indica | Whole |
| WDB control | Min | 0.0 | 0.0 | 0.0 |
| Max | 94.5 | 96.7 | 96.7 | |
| Mean | 58.9 | 69.0 | 62.4 | |
| SD | 25.0 | 26.7 | 26.0 | |
| CV (%) | 42.4 | 38.6 | 41.7 | |
| DH | Min | 3.1 | 0.0 | 0.0 |
| Max | 98.0 | 100.0 | 100.0 | |
| Mean | 67.6 | 71.2 | 69.0 | |
| SD | 23.2 | 32.8 | 27.2 | |
| CV (%) | 34.3 | 46.1 | 39.5 | |
| GA | Min | 2.4 | 2.7 | 2.4 |
| Max | 98.5 | 98.5 | 98.5 | |
| Mean | 73.0 | 74.5 | 73.6 | |
| SD | 21.8 | 29.2 | 24.7 | |
| CV (%) | 29.9 | 39.1 | 33.6 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Zou, W.; Zhang, M.; Liu, J.; Chen, L.; Peng, T.; Ye, G. Genome-Wide Association Study for Seed Dormancy Using Re-Sequenced Germplasm under Multiple Conditions in Rice. Int. J. Mol. Sci. 2023, 24, 6117. https://doi.org/10.3390/ijms24076117
Chen D, Zou W, Zhang M, Liu J, Chen L, Peng T, Ye G. Genome-Wide Association Study for Seed Dormancy Using Re-Sequenced Germplasm under Multiple Conditions in Rice. International Journal of Molecular Sciences. 2023; 24(7):6117. https://doi.org/10.3390/ijms24076117
Chicago/Turabian StyleChen, Dandan, Wenli Zou, Mingpei Zhang, Jindong Liu, Liang Chen, Ting Peng, and Guoyou Ye. 2023. "Genome-Wide Association Study for Seed Dormancy Using Re-Sequenced Germplasm under Multiple Conditions in Rice" International Journal of Molecular Sciences 24, no. 7: 6117. https://doi.org/10.3390/ijms24076117