

Development of Novel Pt(IV)-Carbohydrate Derivatives as Targeted Anticancer Agents against Osteosarcoma
 , ,
, ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterisation
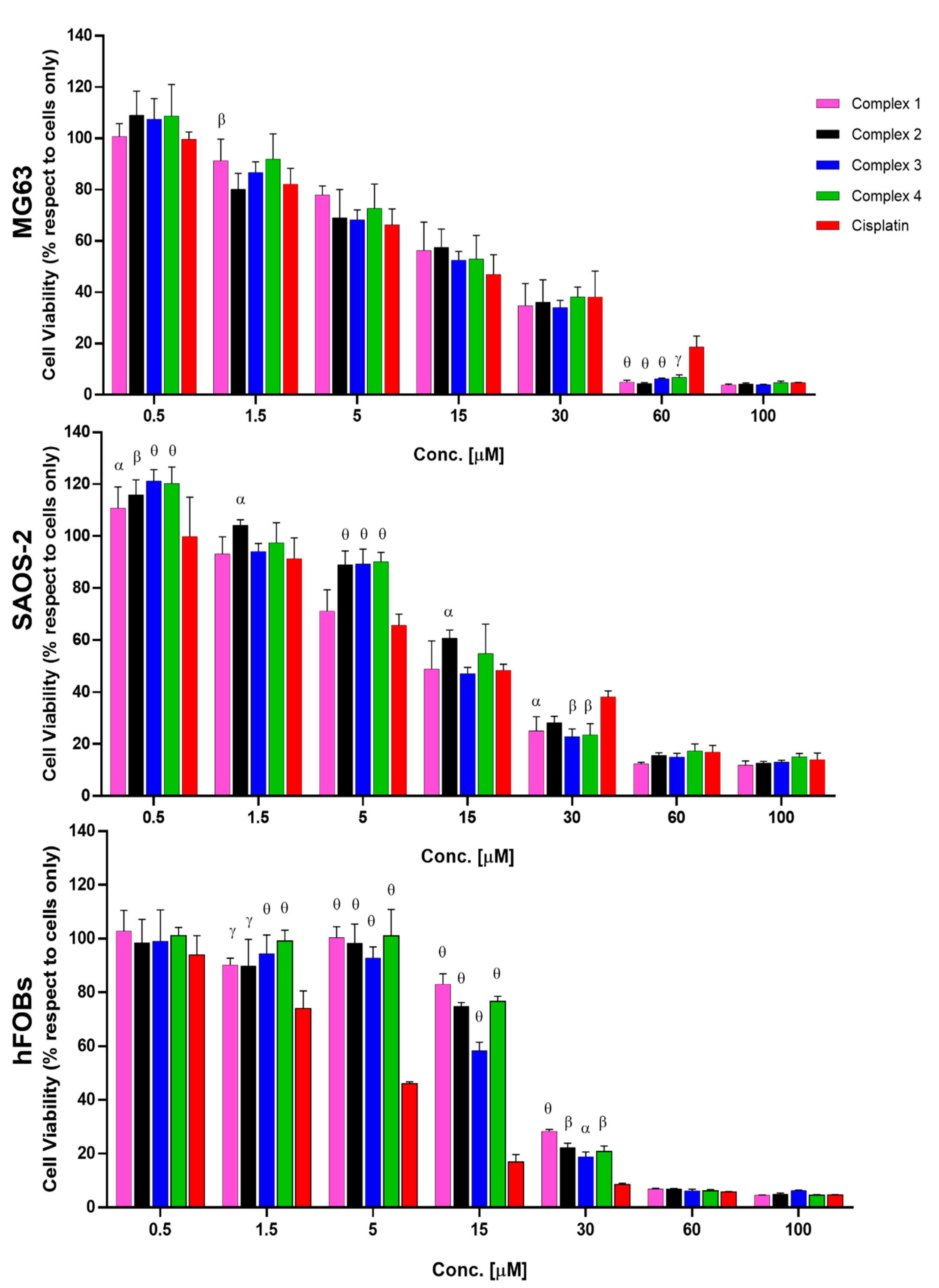
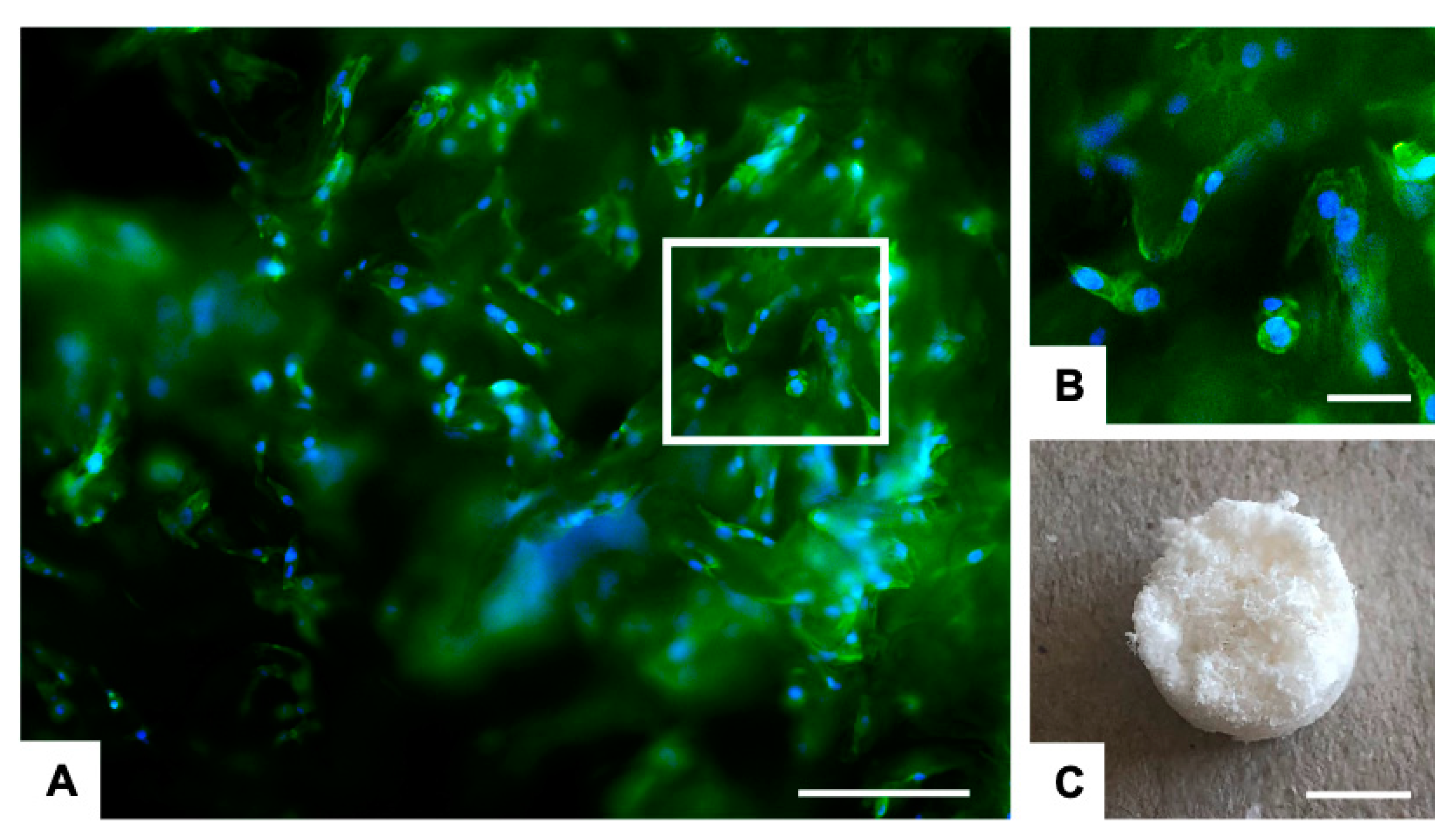
2.2. Biological Evaluation
3. Materials and Methods
3.1. General Methods
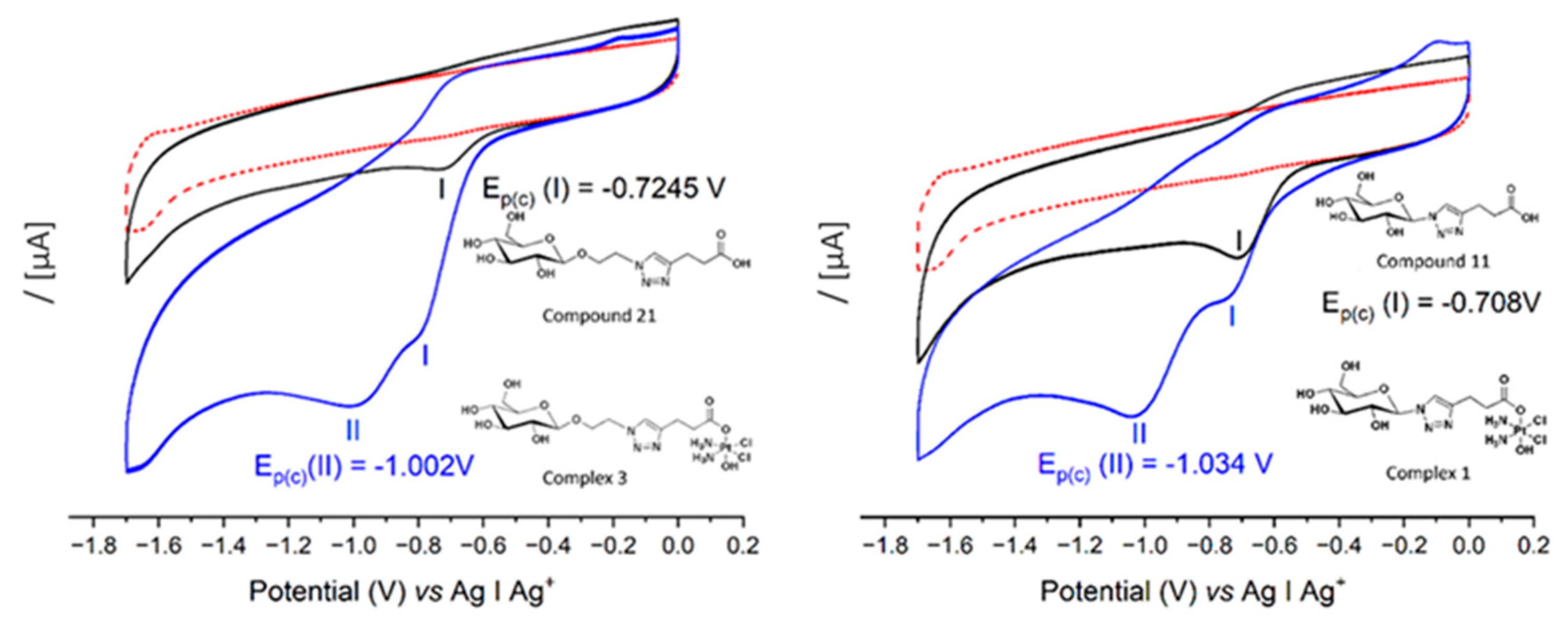
3.2. Cyclic Voltammetry
3.3. Stability and Reduction Studies
3.4. In Vitro Biological Evaluation
3.5. Synthetic Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mortensen, A.C.L.; Mohajershojai, T.; Hariri, M.; Pettersson, M.; Spiegelberg, D. Overcoming Limitations of Cisplatin Therapy by Additional Treatment With the HSP90 Inhibitor Onalespib. Front. Oncol. 2020, 10, 532285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef] [PubMed]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef] [PubMed]
- Fronik, P.; Gutmann, M.; Vician, P.; Stojanovic, M.; Kastner, A.; Heffeter, P.; Pirker, C.; Keppler, B.K.; Berger, W.; Kowol, C.R. A platinum(IV) prodrug strategy to overcome glutathione-based oxaliplatin resistance. Commun. Chem. 2022, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Harrap, K.R.; Kelland, L.R.; Jones, M.; Goddard, P.M.; Orr, R.M.; Morgan, S.E.; Murrer, B.A.; Abrams, M.J.; Giandomenico, C.M.; Cobbleigh, T. Platinum coordination complexes which circumvent cisplatin resistance. Adv. Enzym. Regul. 1991, 31, 31–43. [Google Scholar] [CrossRef]
- Bhargava, A.; Vaishampayan, U.N. Satraplatin: Leading the new generation of oral platinum agents. Expert Opin. Investig. Drugs 2009, 18, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Eastman, A. Glutathione-mediated activation of anticancer platinum(IV) complexes. Biochem. Pharmacol. 1987, 36, 4177–4178. [Google Scholar] [CrossRef]
- Rischin, D.; Ling, V. Ormaplatin resistance is associated with decreased accumulation of its platinum (II) analogue, dichloro(D,L-trans)1,2-diaminocyclohexaneplatinum (II). Br. J. Cancer 1996, 74, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Pendyala, L.; Krishnan, B.S.; Walsh, J.R.; Arakali, A.V.; Cowens, J.W.; Creaven, P.J. Studies on the Human Metabolism of Iproplatin. Cancer Chemother. Pharmacol. 1989, 25, 10–14. [Google Scholar] [CrossRef]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ma, R.; Wang, Y.; Zhu, X.; Zhang, J.; Chan, H.C.; Chen, X.; Zhang, W.; Chiu, S.-K.; Zhu, G. Chalcoplatin, a dual-targeting and p53 activator-containing anticancer platinum(iv) prodrug with unique mode of action. Chem. Commun. 2015, 51, 6301–6304. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, X.; Hao, W.; Huang, Z.; Wang, X.; Wang, P.G. Mono-functionalized glycosylated platinum(IV) complexes possessed both pH and redox dual-responsive properties: Exhibited enhanced safety and preferentially accumulated in cancer cells in vitro and in vivo. Eur. J. Med. Chem. 2017, 128, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, Q.; Huang, Z.; Yang, X.; Nie, Q.; Hao, W.; Wang, P.G.; Wang, X. Glycosylated Platinum(IV) Complexes as Substrates for Glucose Transporters (GLUTs) and Organic Cation Transporters (OCTs) Exhibited Cancer Targeting and Human Serum Albumin Binding Properties for Drug Delivery. J. Med. Chem. 2017, 60, 5736–5748. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Q.; Yang, X.; Hao, W.; Huang, Z.; Zhang, J.; Wang, X.; Wang, P.G. Glycosylated platinum(iv) prodrugs demonstrated significant therapeutic efficacy in cancer cells and minimized side-effects. Dalton Trans. 2016, 45, 11830–11838. [Google Scholar] [CrossRef]
- Ma, J.; Liu, H.; Xi, Z.; Hou, J.; Li, Y.; Niu, J.; Liu, T.; Bi, S.; Wang, X.; Wang, C.; et al. Protected and De-protected Platinum(IV) Glycoconjugates With GLUT1 and OCT2-Mediated Selective Cancer Targeting: Demonstrated Enhanced Transporter-Mediated Cytotoxic Properties in vitro and in vivo. Front. Chem. 2018, 6, 386. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Huang, Z.; Ma, J.; Lu, X.; Zhang, L.; Wang, X.; Wang, P.G. Design, synthesis and biological evaluation of a novel series of glycosylated platinum(iv) complexes as antitumor agents. Dalton Trans. 2016, 45, 10366–10374. [Google Scholar] [CrossRef]
- Heymann, M.-F.; Lézot, F.; Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell. Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef]
- Li, C.; Cai, J.; Ge, F.; Wang, G. TGM2 knockdown reverses cisplatin chemoresistance in osteosarcoma. Int. J. Mol. Med. 2018, 42, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynihan, E.; Bassi, G.; Ruffini, A.; Panseri, S.; Montesi, M.; Velasco-Torrijos, T.; Montagner, D. Click Pt(IV)-Carbohydrates Pro-Drugs for Treatment of Osteosarcoma. Front. Chem. 2021, 9, 795997. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, K.; Hamidullah, H.; Singh, K.; Arun, A.; Shukla, M.; Srivastava, N.; Ashraf, R.; Sharma, A.; Mahar, R.; Shukla, S.K.; et al. Identification of gallic acid based glycoconjugates as a novel tubulin polymerization inhibitors. Org. Biomol. Chem. 2015, 14, 1338–1358. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zeng, X.; Yan, C.; Sun, X.; Gong, X.; Rao, Y.; Yan, N. Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature 2012, 490, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Holman, G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflug. Arch.-Eur. J. Physiol. 2020, 472, 1155–1175. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.S.; Ahmed, S.A.; Althagafi, I.I.; Ansari, M.A.; Kamal, A. Application of triazoles as bioisosteres and linkers in the development of microtubule targeting agents. RSC Med. Chem. 2020, 11, 327–348. [Google Scholar] [CrossRef]
- Liu, X.; Barth, M.; Cseh, K.; Kowol, C.R.; Jakupec, M.A.; Keppler, B.K.; Gibson, D.; Weigand, W. Oxoplatin-Based Pt(IV) Lipoate Complexes and Their Biological Activity. Chem. Biodivers. 2022, 19, e202200695. [Google Scholar] [CrossRef]
- Chen, C.K.J.; Kappen, P.; Gibson, D.; Hambley, T.W. trans-Platinum(iv) pro-drugs that exhibit unusual resistance to reduction by endogenous reductants and blood serum but are rapidly activated inside cells:1H NMR and XANES spectroscopy study. Dalton Trans. 2020, 49, 7722–7736. [Google Scholar] [CrossRef]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef]
- Cifuentes, M.; García, M.A.; Arrabal, P.M.; Martínez, F.; Yañez, M.J.; Jara, N.; Weil, B.; Domínguez, D.; Medina, R.A.; Nualart, F. Insulin regulates GLUT1-mediated glucose transport in MG-63 human osteosarcoma cells. J. Cell. Physiol. 2011, 226, 1425–1432. [Google Scholar] [CrossRef]
- Arponen, M.; Jalava, N.; Widjaja, N.; Ivaska, K.K. Glucose transporters GLUT1, GLUT3, and GLUT4 have different effects on osteoblast proliferation and metabolism. Front. Physiol. 2022, 13, 1035516. [Google Scholar] [CrossRef]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Del Nery, E.; Ebner, D.; Montoya, M.C.; Östling, P.; Pietiäinen, V.; Price, L.S.; et al. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. ASSAY Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, G.S.; Gostynska, N.; Dapporto, M.; Campodoni, E.; Montesi, M.; Panseri, S.; Tampieri, A.; Kon, E.; Marcacci, M.; Sprio, S.; et al. Evaluation of different crosslinking agents on hybrid biomimetic collagen-hydroxyapatite composites for regenerative medicine. Int. J. Biol. Macromol. 2018, 106, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Bassi, G.; Panseri, S.; Dozio, S.M.; Sandri, M.; Campodoni, E.; Dapporto, M.; Sprio, S.; Tampieri, A.; Montesi, M. Scaffold-based 3D cellular models mimicking the heterogeneity of osteosarcoma stem cell niche. Sci. Rep. 2020, 10, 22294. [Google Scholar] [CrossRef] [PubMed]
- Dozio, S.M.; Montesi, M.; Campodoni, E.; Sandri, M.; Piattelli, A.; Tampieri, A.; Panseri, S. Differences in osteogenic induction of human mesenchymal stem cells between a tailored 3D hybrid scaffold and a 2D standard culture. J. Mater. Sci. Mater. Med. 2019, 30, 136. [Google Scholar] [CrossRef]
- Dhara, S.C. A Rapid Method for the Synthesis of Cis-[Pt(NH3)2Cl2]. Indian J. Chem. 1970, 8, 193–194. [Google Scholar]
- Brandon, R.J.; Dabrowiak, J.C. Synthesis, characterization, and properties, of a group of platinum(IV) complexes. J. Med. Chem. 1984, 27, 861–865. [Google Scholar] [CrossRef]
- Valle, S.; Alcalá, S.; Martin-Hijano, L.; Cabezas-Sáinz, P.; Navarro, D.; Muñoz, E.R.; Yuste, L.; Tiwary, K.; Walter, K.; Ruiz-Cañas, L.; et al. Exploiting oxidative phosphorylation to promote the stem and immunoevasive properties of pancreatic cancer stem cells. Nat. Commun. 2020, 11, 5265. [Google Scholar] [CrossRef]
- Zheng, D.; Sussman, J.H.; Jeon, M.P.; Parrish, S.T.; MacMullan, M.A.; Delfarah, A.; Graham, N.A. AKT but not MYC promotes reactive oxygen species-mediated cell death in oxidative culture. J. Cell Sci. 2020, 133, jcs239277. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.; Awuah, S.G.; Lippard, S.J. Chemical Approach to Positional Isomers of Glucose–Platinum Conjugates Reveals Specific Cancer Targeting through Glucose-Transporter-Mediated Uptake in Vitro and in Vivo. J. Am. Chem. Soc. 2016, 138, 12541–12551. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cisplatin | Complex 1 | Complex 2 | Complex 3 | Complex 4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 95% CI | 95% CI | 95% CI | 95% CI | 95% CI | ||||||
| MG63 | 12.20 | +3.1; −2.6 | 15.90 | +3.3; −2.9 | 13.60 | +5.2; −4.0 | 13.0 | +3.2; −2.7 | 14.90 | +4.2; −3.5 |
| SAOS-2 | 13.70 | +3.2; −2.7 | 12.94 | +3.4; −3.0 | 19.09 | +3.4; −3.0 | 15.18 | +3.7; −3.1 | 17.22 | +4.5; −3.7 |
| hFOBs | 4.109 | +0.42; −0.47 | 23.36 | +1.72; −1.82 | 20.85 | +1.92; −2.11 | 16.88 | +1.7; −1.75 | 20.91 | +0.42; −0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moynihan, E.; Panseri, S.; Bassi, G.; Rossi, A.; Campodoni, E.; Dempsey, E.; Montesi, M.; Velasco-Torrijos, T.; Montagner, D. Development of Novel Pt(IV)-Carbohydrate Derivatives as Targeted Anticancer Agents against Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 6028. https://doi.org/10.3390/ijms24076028
Moynihan E, Panseri S, Bassi G, Rossi A, Campodoni E, Dempsey E, Montesi M, Velasco-Torrijos T, Montagner D. Development of Novel Pt(IV)-Carbohydrate Derivatives as Targeted Anticancer Agents against Osteosarcoma. International Journal of Molecular Sciences. 2023; 24(7):6028. https://doi.org/10.3390/ijms24076028
Chicago/Turabian StyleMoynihan, Eoin, Silvia Panseri, Giada Bassi, Arianna Rossi, Elisabetta Campodoni, Eithne Dempsey, Monica Montesi, Trinidad Velasco-Torrijos, and Diego Montagner. 2023. "Development of Novel Pt(IV)-Carbohydrate Derivatives as Targeted Anticancer Agents against Osteosarcoma" International Journal of Molecular Sciences 24, no. 7: 6028. https://doi.org/10.3390/ijms24076028