
Neuropharmacological Modulation of N-methyl-D-aspartate, Noradrenaline and Endocannabinoid Receptors in Fear Extinction Learning: Synaptic Transmission and Plasticity




Abstract
:1. Introduction
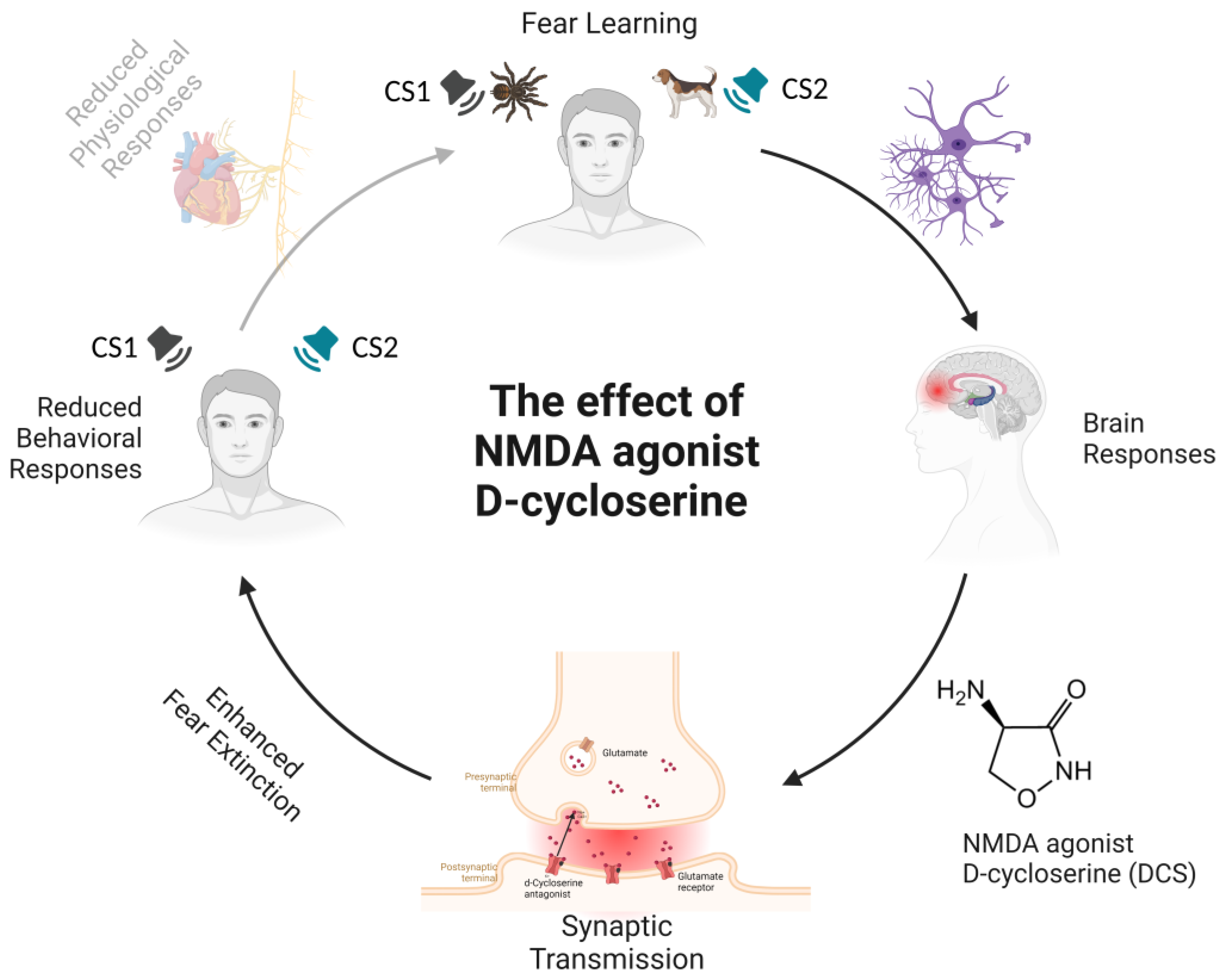
2. The Effects of NMDA Agonist D-Cycloserine (DCS) and Valproic Acid (VPA)
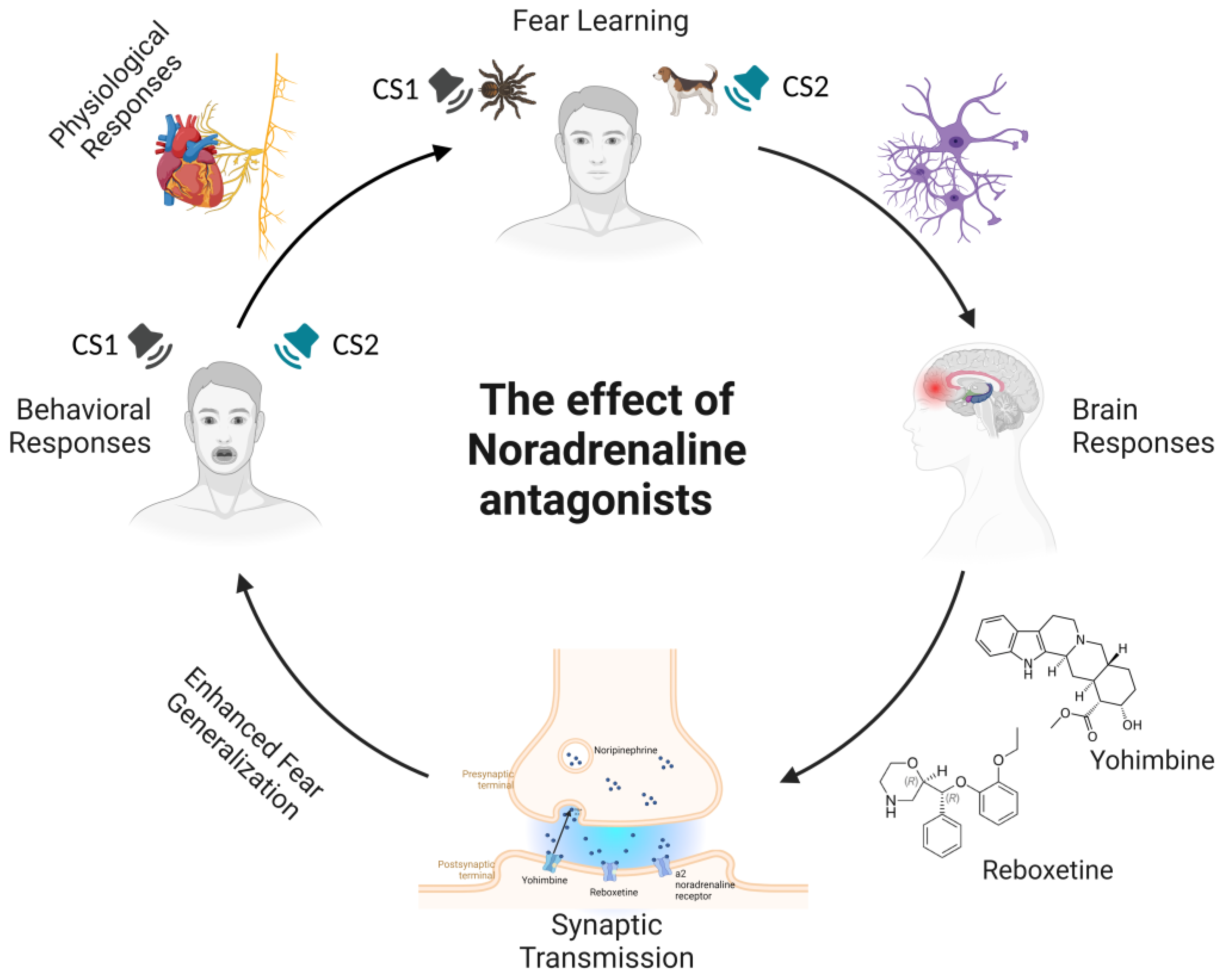
3. Effects of Noradrenaline (NA) Modulation
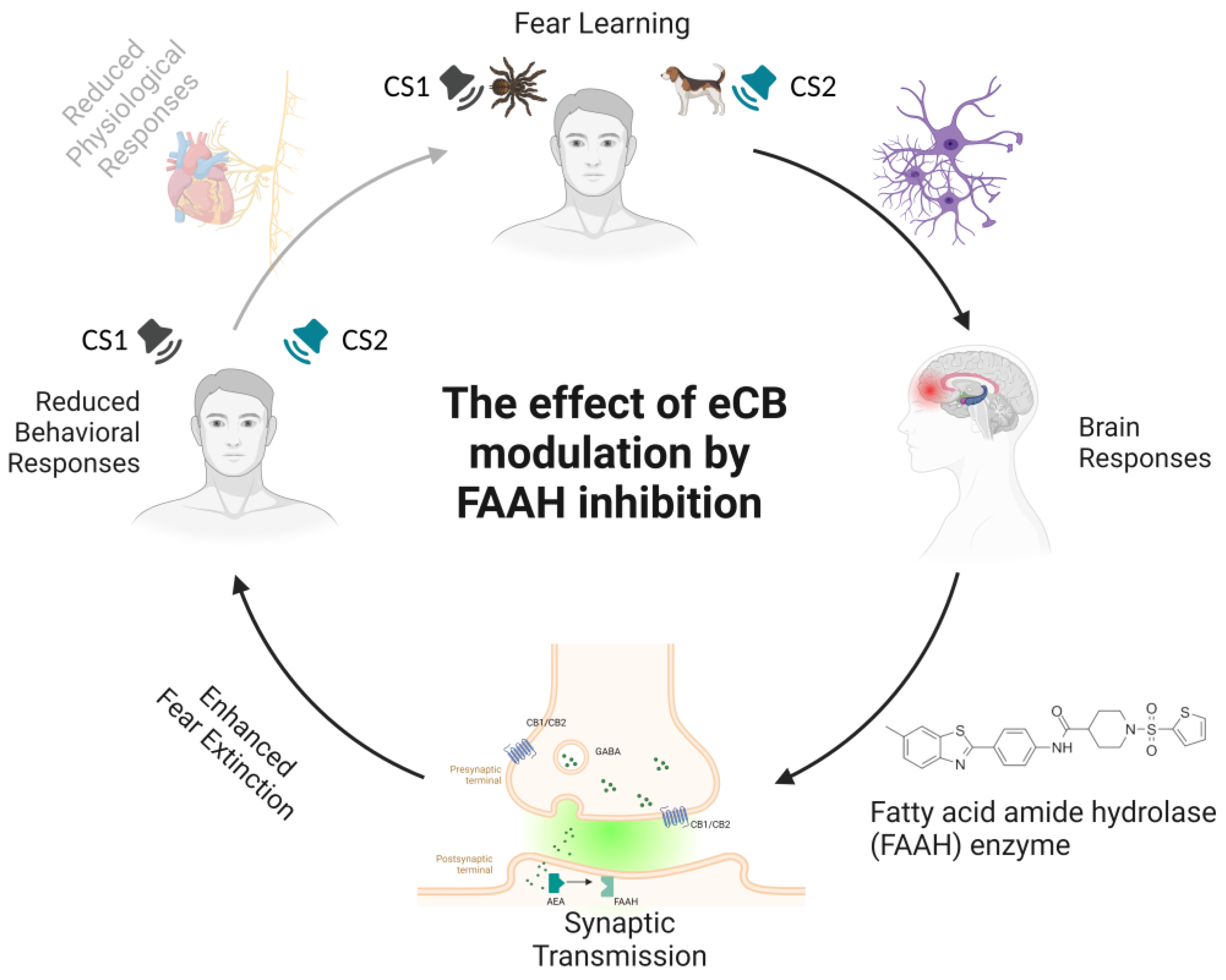
4. The Effects of Fatty Acid Amide Hydrolase (FAAH) Inhibition
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NS | neutral stimulus |
| US | unconditioned stimulus |
| CS+ | conditioned stimulus |
| CS- | conditioned stimulus never paired with the US |
| CXT | conditioned context |
| CR | conditioned response |
| SCR | skin conductance response |
| EMG | electromyography |
| fMRI | functional magnetic resonance imaging |
| BOLD | blood-oxygen-level-dependent |
| PTSD | post-traumatic stress disorder |
| NMDA | N-methyl-D-aspartate |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionate |
| DCS | D-cycloserine |
| VPA | valproic acid |
| NA | noradrenaline |
| YOH | yohimbine |
| RBX | reboxetine |
| eCBs | endocannabinoids |
| AEA | anandamide |
| FAAH | fatty acid amide hydrolase |
| FAAHi | FAAH inhibitor |
| MAGL | monoacylglycerol lipase |
References
- Bouton, M.E. Context and Behavioral Processes in Extinction. Learn. Mem. 2004, 11, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechara, A.; Damasio, H.; Damasio, A.R.; Lee, G.P. Different Contributions of the Human Amygdala and Ventromedial Prefrontal Cortex to Decision-Making. J. Neurosci. 1999, 19, 5473–5481. [Google Scholar] [CrossRef] [Green Version]
- Lonsdorf, T.B.; Menz, M.M.; Andreatta, M.; Fullana, M.A.; Golkar, A.; Haaker, J.; Heitland, I.; Hermann, A.; Kuhn, M.; Kruse, O.; et al. Don’t Fear ‘Fear Conditioning’: Methodological Considerations for the Design and Analysis of Studies on Human Fear Acquisition, Extinction, and Return of Fear. Neurosci. Biobehav. Rev. 2017, 77, 247–285. [Google Scholar] [CrossRef]
- Maren, S.; Quirk, G.J. Neuronal Signalling of Fear Memory. Nat. Rev. Neurosci. 2004, 5, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Quirk, G.J.; Mueller, D. Neural Mechanisms of Extinction Learning and Retrieval. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Borgomaneri, S.; Battaglia, S.; Garofalo, S.; Tortora, F.; Avenanti, A.; di Pellegrino, G. State-Dependent TMS over Prefrontal Cortex Disrupts Fear-Memory Reconsolidation and Prevents the Return of Fear. Curr. Biol. 2020, 30, 3672–3679.e4. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. The Influence of Vicarious Fear-Learning in “Infecting” Reactive Action Inhibition. Front. Behav. Neurosci. 2022, 16, 946263. [Google Scholar] [CrossRef]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the Human Ventromedial Prefrontal Cortex Support Fear Learning, Fear Extinction or Both? A Commentary on Subregional Contributions. Mol. Psychiatry 2021, 27, 784–786. [Google Scholar] [CrossRef]
- Fullana, M.A.; Harrison, B.J.; Soriano-Mas, C.; Vervliet, B.; Cardoner, N.; Àvila-Parcet, A.; Radua, J. Neural Signatures of Human Fear Conditioning: An Updated and Extended Meta-Analysis of FMRI Studies. Mol. Psychiatry 2016, 21, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Haaker, J.; Lonsdorf, T.B.; Schümann, D.; Menz, M.; Brassen, S.; Bunzeck, N.; Gamer, M.; Kalisch, R. Deficient Inhibitory Processing in Trait Anxiety: Evidence from Context-Dependent Fear Learning, Extinction Recall and Renewal. Biol. Psychol. 2015, 111, 65–72. [Google Scholar] [CrossRef]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing Cardiac Autonomic Dynamics of Fear Learning in Humans. Psychophysiology 2022, 59, e14122. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, C.; Grasso, G.M.; Nitsche, M.A.; D’Italia, G.; Sortino, M.; Salehinejad, M.A.; Falzone, A.; Avenanti, A.; Vicario, C.M. Enhanced Fear Acquisition in Individuals with Evening Chronotype. A Virtual Reality Fear Conditioning/Extinction Study. J. Affect. Disord. 2022, 311, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Vicario, C.M.; Nitsche, M.A.; Hoysted, I.; Yavari, F.; Avenanti, A.; Salehinejad, M.A.; Felmingham, K.L. Anodal Transcranial Direct Current Stimulation over the Ventromedial Prefrontal Cortex Enhances Fear Extinction in Healthy Humans: A Single Blind Sham-Controlled Study. Brain Stimul. 2020, 13, 489–491. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. Stopping in (e)Motion: Reactive Action Inhibition When Facing Valence-Independent Emotional Stimuli. Front. Behav. Neurosci. 2022, 16, 998714. [Google Scholar] [CrossRef]
- Campbell, T.L.; Kochli, D.E.; McDaniel, M.A.; Myers, M.K.; Dunn, M.E.; Diana, V.A.; Quinn, J.J. Using Extinction-Renewal to Circumvent the Memory Strength Boundary Condition in Fear Memory Reconsolidation. Brain Sci. 2021, 11, 1023. [Google Scholar] [CrossRef] [PubMed]
- Etkin, A.; Egner, T.; Kalisch, R. Emotional Processing in Anterior Cingulate and Medial Prefrontal Cortex. Trends Cogn. Sci. 2011, 15, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Alberini, C.M.; Ledoux, J.E. Memory Reconsolidation. Curr. Biol. 2013, 23, R746–R750. [Google Scholar] [CrossRef] [Green Version]
- Kindt, M.; Soeter, M.; Vervliet, B. Beyond Extinction: Erasing Human Fear Responses and Preventing the Return of Fear. Nat. Neurosci. 2009, 12, 256–258. [Google Scholar] [CrossRef]
- Nader, K.; Hardt, O. A Single Standard for Memory: The Case for Reconsolidation. Nat. Rev. Neurosci. 2009, 10, 224–234. [Google Scholar] [CrossRef]
- Bouton, M.E.; King, D.A. Contextual Control of the Extinction of Conditioned Fear: Tests for the Associative Value of the Context. J. Exp. Psychol. Anim. Behav. Process. 1983, 9, 248–265. [Google Scholar] [CrossRef]
- Battaglia, S. Neurobiological Advances of Learned Fear in Humans. Adv. Clin. Exp. Med. 2022, 31, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Kalisch, R.; Korenfeld, E.; Stephan, K.E.; Weiskopf, N.; Seymour, B.; Dolan, R.J. Context-Dependent Human Extinction Memory Is Mediated by a Ventromedial Prefrontal and Hippocampal Network. J. Neurosci. 2006, 26, 9503–9511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taschereau-Dumouchel, V.; Kawato, M.; Lau, H. Multivoxel Pattern Analysis Reveals Dissociations between Subjective Fear and Its Physiological Correlates. Mol. Psychiatry 2020, 25, 2342–2354. [Google Scholar] [CrossRef] [Green Version]
- Lonsdorf, T.B.; Richter, J. Challenges of Fear Conditioning Research in the Age of RDoC. J. Psychol. 2017, 225, 189–199. [Google Scholar] [CrossRef]
- Myers, K.M.; Davis, M. Mechanisms of Fear Extinction. Mol. Psychiatry 2007, 12, 120–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raij, T.; Nummenmaa, A.; Marin, M.F.; Porter, D.; Furtak, S.; Setsompop, K.; Milad, M.R. Prefrontal Cortex Stimulation Enhances Fear Extinction Memory in Humans. Biol. Psychiatry 2018, 84, 129–137. [Google Scholar] [CrossRef]
- Milad, M.R.; Quirk, G.J. Fear Extinction as a Model for Translational Neuroscience: Ten Years of Progress. Annu. Rev. Psychol. 2012, 63, 129–151. [Google Scholar] [CrossRef] [Green Version]
- Harrison, B.J.; Fullana, M.A.; Via, E.; Soriano-Mas, C.; Vervliet, B.; Martínez-Zalacaín, I.; Pujol, J.; Davey, C.G.; Kircher, T.; Straube, B.; et al. Human Ventromedial Prefrontal Cortex and the Positive Affective Processing of Safety Signals. Neuroimage 2017, 152, 12–18. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Sciamanna, G.; Tortora, F.; Laricchiuta, D. Memories Are Not Written in Stone: Re-Writing Fear Memories by Means of Non-Invasive Brain Stimulation and Optogenetic Manipulations. Neurosci. Biobehav. Rev. 2021, 127, 334–352. [Google Scholar] [CrossRef]
- Marković, V.; Vicario, C.M.; Yavari, F.; Salehinejad, M.A.; Nitsche, M.A. A Systematic Review on the Effect of Transcranial Direct Current and Magnetic Stimulation on Fear Memory and Extinction. Front. Hum. Neurosci. 2021, 15, 655947. [Google Scholar] [CrossRef]
- Cahill, E.N.; Milton, A.L. Neurochemical and Molecular Mechanisms Underlying the Retrieval-Extinction Effect. Psychopharmacology 2019, 236, 111–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsicano, G.; Wotjak, C.T.; Azad, S.C.; Bisogno, T.; Rammes, G.; Cascio, M.G.; Hermann, H.; Tang, J.; Hofmann, C.; Zieglgänsberger, W.; et al. The Endogenous Cannabinoid System Controls Extinction of Aversive Memories. Nature 2002, 418, 530–534. [Google Scholar] [CrossRef]
- de Bitencourt, R.M.; Pamplona, F.A.; Takahashi, R.N. A Current Overview of Cannabinoids and Glucocorticoids in Facilitating Extinction of Aversive Memories: Potential Extinction Enhancers. Neuropharmacology 2013, 64, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Steckler, T.; Risbrough, V. Pharmacological Treatment of PTSD—Established and New Approaches. Neuropharmacology 2012, 62, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Thayer, J.F.; Lane, R. Perseverative Thinking and Health: Neurovisceral Concomitants. Psychol. Health 2002, 17, 685–695. [Google Scholar] [CrossRef]
- Thayer, J.F.; Siegle, G.J. Neurovisceral Integration in Cardiac and Emotional Regulation. IEEE Eng. Med. Biol. Mag. 2002, 21, 24–29. [Google Scholar] [CrossRef]
- Battaglia, S.; Thayer, J.F. Functional Interplay between Central and Autonomic Nervous Systems in Human Fear Conditioning. Trends Neurosci. 2022, 45, 4. [Google Scholar] [CrossRef]
- Moreira, F.A.; Wotjak, C.T. Cannabinoids and Anxiety. Curr. Top. Behav. Neurosci. 2010, 2, 429–450. [Google Scholar] [CrossRef]
- Lafenêtre, P.; Chaouloff, F.; Marsicano, G. The Endocannabinoid System in the Processing of Anxiety and Fear and How CB1 Receptors May Modulate Fear Extinction. Pharmacol. Res. 2007, 56, 367–381. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Fanselow, M.S.; Ledoux, J.E. Why We Think Pavlovian Fear Conditioning Occurs in the Basolateral Amygdala. Neuron 1999, 23, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanselow, M.S.; Poulos, A.M. The Neuroscience of Mammalian Associative Learning. Annu. Rev. Psychol. 2005, 56, 207–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, E.; Ge, H.; Ren, K.; Pena de Ortiz, S.; Quirk, G.J. Consolidation of Fear Extinction Requires Protein Synthesis in the Medial Prefrontal Cortex. J. Neurosci. 2004, 24, 5704–5710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G.; Starita, F. Revaluing the Role of VmPFC in the Acquisition of Pavlovian Threat Conditioning in Humans. J. Neurosci. 2020, 40, 8491–8500. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef] [Green Version]
- Martos, D.; Tuka, B.; Tanaka, M.; Vécsei, L.; Telegdy, G. Memory Enhancement with Kynurenic Acid and Its Mechanisms in Neurotransmission. Biomedicines 2022, 10, 849. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Lester, R.A. Excitatory Amino Acid Receptors in the Vertebrate Central Nervous System. Pharmacol. Rev. 1989, 41, 143–210. [Google Scholar]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [Green Version]
- Bahn, S.; Volk, B.; Wisden, W. Kainate Receptor Gene Expression in the Developing Rat Brain. J. Neurosci. Off. J. Soc. Neurosci. 1994, 14, 5525–5547. [Google Scholar] [CrossRef] [PubMed]
- Egebjerg, J.; Bettler, B.; Hermans-Borgmeyer, I.; Heinemann, S. Cloning of a CDNA for a Glutamate Receptor Subunit Activated by Kainate but Not AMPA. Nature 1991, 351, 745–748. [Google Scholar] [CrossRef]
- Bloss, E.B.; Hunter, R.G. Hippocampal Kainate Receptors. Vitam. Horm. 2010, 82, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.; Zhao, M.-G.; Toyoda, H.; Qiu, C.-S.; Zhuo, M. Altered Behavioral Responses to Noxious Stimuli and Fear in Glutamate Receptor 5 (GluR5)- or GluR6-Deficient Mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 977–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.; Hillman, B.G.; Gupta, S.C.; Suryavanshi, P.; Bhatt, J.M.; Pavuluri, R.; Stairs, D.J.; Dravid, S.M. Deletion of Glutamate Delta-1 Receptor in Mouse Leads to Enhanced Working Memory and Deficit in Fear Conditioning. PLoS ONE 2013, 8, e60785. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.J.; Liu, S.J. GluN2D NMDA Receptors Gate Fear Extinction Learning and Interneuron Plasticity. Front. Synaptic Neurosci. 2021, 13, 681068. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; Bhat, J.A.; Satti, N.K.; Sharma, P.R.; Hamid, A.; Ahmad, M. Withanone, an Active Constituent from Withania Somnifera, Affords Protection Against NMDA-Induced Excitotoxicity in Neuron-like Cells. Mol. Neurobiol. 2017, 54, 5061–5073. [Google Scholar] [CrossRef] [PubMed]
- Myers-Schulz, B.; Koenigs, M. Functional Anatomy of Ventromedial Prefrontal Cortex: Implications for Mood and Anxiety Disorders. Mol. Psychiatry 2012, 17, 132–141. [Google Scholar] [CrossRef]
- Matsumoto, T.; Rauskolb, S.; Polack, M.; Klose, J.; Kolbeck, R.; Korte, M.; Barde, Y.-A. Biosynthesis and Processing of Endogenous BDNF: CNS Neurons Store and Secrete BDNF, Not pro-BDNF. Nat. Neurosci. 2008, 11, 131–133. [Google Scholar] [CrossRef]
- Yamamoto, S.; Morinobu, S.; Fuchikami, M.; Kurata, A.; Kozuru, T.; Yamawaki, S. Effects of Single Prolonged Stress and D-Cycloserine on Contextual Fear Extinction and Hippocampal NMDA Receptor Expression in a Rat Model of PTSD. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 2108–2116. [Google Scholar] [CrossRef] [Green Version]
- Myers, K.M.; Carlezon, W.A.J.; Davis, M. Glutamate Receptors in Extinction and Extinction-Based Therapies for Psychiatric Illness. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2011, 36, 274–293. [Google Scholar] [CrossRef]
- Ressler, K.J.; Rothbaum, B.O.; Tannenbaum, L.; Anderson, P.; Graap, K.; Zimand, E.; Hodges, L.; Davis, M. Cognitive Enhancers as Adjuncts to Psychotherapy: Use of D-Cycloserine in Phobic Individuals to Facilitate Extinction of Fear. Arch. Gen. Psychiatry 2004, 61, 1136–1144. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Avenanti, A.; di Pellegrino, G. Don’t Hurt Me No More: State-Dependent Transcranial Magnetic Stimulation for the Treatment of Specific Phobia. J. Affect. Disord. 2021, 286, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Guastella, A.J.; Richardson, R.; Lovibond, P.F.; Rapee, R.M.; Gaston, J.E.; Mitchell, P.; Dadds, M.R. A Randomized Controlled Trial of D-Cycloserine Enhancement of Exposure Therapy for Social Anxiety Disorder. Biol. Psychiatry 2008, 63, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.W.; Tolin, D.F.; Simon, N.M.; Pearlson, G.D.; Basden, S.; Meunier, S.A.; Hofmann, S.G.; Eisenmenger, K.; Krystal, J.H.; Pollack, M.H. Efficacy of D-Cycloserine for Enhancing Response to Cognitive-Behavior Therapy for Panic Disorder. Biol. Psychiatry 2010, 67, 365–370. [Google Scholar] [CrossRef]
- Storch, E.A.; Larson, M.J.; Muroff, J.; Caporino, N.; Geller, D.; Reid, J.M.; Morgan, J.; Jordan, P.; Murphy, T.K. Predictors of Functional Impairment in Pediatric Obsessive-Compulsive Disorder. J. Anxiety Disord. 2010, 24, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Farzam, K.; Kidron, A.; Lakhkar, A.D. Adrenergic Drugs; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Alhayek, S.; Preuss, C.V. Beta 1 Receptors; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Braga, M.F.M.; Aroniadou-Anderjaska, V.; Manion, S.T.; Hough, C.J.; Li, H. Stress Impairs Alpha(1A) Adrenoceptor-Mediated Noradrenergic Facilitation of GABAergic Transmission in the Basolateral Amygdala. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2004, 29, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Do-Monte, F.H.; Manzano-Nieves, G.; Quiñones-Laracuente, K.; Ramos-Medina, L.; Quirk, G.J. Revisiting the Role of Infralimbic Cortex in Fear Extinction with Optogenetics. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 3607–3615. [Google Scholar] [CrossRef] [Green Version]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic Monoacylglycerol Lipase Blockade Causes Functional Antagonism of the Endocannabinoid System. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira Alvares, L.; de Oliveira, L.F.; Camboim, C.; Diehl, F.; Genro, B.P.; Lanziotti, V.B.; Quillfeldt, J.A. Amnestic Effect of Intrahippocampal AM251, a CB1-Selective Blocker, in the Inhibitory Avoidance, but Not in the Open Field Habituation Task, in Rats. Neurobiol. Learn. Mem. 2005, 83, 119–124. [Google Scholar] [CrossRef]
- Kinsey, S.G.; Mahadevan, A.; Zhao, B.; Sun, H.; Naidu, P.S.; Razdan, R.K.; Selley, D.E.; Imad Damaj, M.; Lichtman, A.H. The CB2 Cannabinoid Receptor-Selective Agonist O-3223 Reduces Pain and Inflammation without Apparent Cannabinoid Behavioral Effects. Neuropharmacology 2011, 60, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Spradley, J.M.; Guindon, J.; Hohmann, A.G. Inhibitors of Monoacylglycerol Lipase, Fatty-Acid Amide Hydrolase and Endocannabinoid Transport Differentially Suppress Capsaicin-Induced Behavioral Sensitization through Peripheral Endocannabinoid Mechanisms. Pharmacol. Res. 2010, 62, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Spohrs, J.; Ulrich, M.; Grön, G.; Prost, M.; Plener, P.L.; Fegert, J.M.; Bindila, L.; Abler, B. Fear Extinction Learning and Anandamide: An FMRI Study in Healthy Humans. Transl. Psychiatry 2021, 11, 161. [Google Scholar] [CrossRef] [PubMed]
- Ney, L.J.; Matthews, A.; Bruno, R.; Felmingham, K.L. Cannabinoid Interventions for PTSD: Where to Next? Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 93, 124–140. [Google Scholar] [CrossRef]
- Ney, L.J.; Matthews, A.; Bruno, R.; Felmingham, K.L. Modulation of the Endocannabinoid System by Sex Hormones: Implications for Posttraumatic Stress Disorder. Neurosci. Biobehav. Rev. 2018, 94, 302–320. [Google Scholar] [CrossRef] [PubMed]
- Kalisch, R.; Gerlicher, A.M.V.; Duvarci, S. A Dopaminergic Basis for Fear Extinction. Trends Cogn. Sci. 2019, 23, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, J.; Ren, L.Y.; Gao, C. N-Methyl D-Aspartate Receptor Subunit Signaling in Fear Extinction. Psychopharmacology 2019, 236, 239–250. [Google Scholar] [CrossRef]
- Warren, W.G.; Papagianni, E.P.; Stevenson, C.W.; Stubbendorff, C. In It Together? The Case for Endocannabinoid-Noradrenergic Interactions in Fear Extinction. Eur. J. Neurosci. 2022, 55, 952–970. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.L.; Davis, M. The Role of Amygdala Glutamate Receptors in Fear Learning, Fear-Potentiated Startle, and Extinction. Pharmacol. Biochem. Behav. 2002, 71, 379–392. [Google Scholar] [CrossRef]
- Gunduz-Cinar, O. The Endocannabinoid System in the Amygdala and Modulation of Fear. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 105, 110116. [Google Scholar] [CrossRef]
- Schiff, H.C.; Johansen, J.P.; Hou, M.; Bush, D.E.A.; Smith, E.K.; Klein, J.E.; LeDoux, J.E.; Sears, R.M. β-Adrenergic Receptors Regulate the Acquisition and Consolidation Phases of Aversive Memory Formation Through Distinct, Temporally Regulated Signaling Pathways. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2017, 42, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, S. Early β Adrenoceptor Dependent Time Window for Fear Memory Persistence in APPswe/PS1dE9 Mice. Sci. Rep. 2021, 11, 870. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Porter, J.T.; Quirk, G.J. Noradrenergic Signaling in Infralimbic Cortex Increases Cell Excitability and Strengthens Memory for Fear Extinction. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustino, T.F.; Maren, S. Noradrenergic Modulation of Fear Conditioning and Extinction. Front. Behav. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Rezvani, A.H. Involvement of the NMDA System in Learning and Memory; Levin, E.D., Buccafusco, J.J., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Walker, D.L.; Davis, M. Are Fear Memories Made and Maintained by the Same NMDA Receptor-Dependent Mechanisms? Neuron 2004, 41, 680–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-C.; Cheng, P.-Y.; Hsiao, M.; Liu, Y.-P. Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. Int. J. Mol. Sci. 2022, 23, 5494. [Google Scholar] [CrossRef]
- Jo, Y.S.; Choi, J.-S. Memory Retrieval in Response to Partial Cues Requires NMDA Receptor-Dependent Neurotransmission in the Medial Prefrontal Cortex. Neurobiol. Learn. Mem. 2014, 109, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Langton, J.M.; Richardson, R. D-Cycloserine Facilitates Extinction the First Time but Not the Second Time: An Examination of the Role of NMDA across the Course of Repeated Extinction Sessions. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 3096–3102. [Google Scholar] [CrossRef]
- Norberg, M.M.; Krystal, J.H.; Tolin, D.F. A Meta-Analysis of D-Cycloserine and the Facilitation of Fear Extinction and Exposure Therapy. Biol. Psychiatry 2008, 63, 1118–1126. [Google Scholar] [CrossRef]
- Fang, A.; Wilhelm, S. Antidepressant Use with D-Cycloserine May Block Fear Extinction. Evid. Based Ment. Health 2016, 19, e5. [Google Scholar] [CrossRef]
- Hofmann, S.G.; Wu, J.Q.; Boettcher, H. D-Cycloserine as an Augmentation Strategy for Cognitive Behavioral Therapy of Anxiety Disorders. Biol. Mood Anxiety Disord. 2013, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- van Berckel, B.N.; Evenblij, C.N.; van Loon, B.J.; Maas, M.F.; van der Geld, M.A.; Wynne, H.J.; van Ree, J.M.; Kahn, R.S. D-Cycloserine Increases Positive Symptoms in Chronic Schizophrenic Patients When Administered in Addition to Antipsychotics: A Double-Blind, Parallel, Placebo-Controlled Study. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 21, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Davis, M. NMDA Receptors and Fear Extinction: Implications for Cognitive Behavioral Therapy. Dialogues Clin. Neurosci. 2011, 13, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; D’Souza, D.C.; Petrakis, I.L.; Belger, A.; Berman, R.M.; Charney, D.S.; Abi-Saab, W.; Madonick, S. NMDA Agonists and Antagonists as Probes of Glutamatergic Dysfunction and Pharmacotherapies in Neuropsychiatric Disorders. Harv. Rev. Psychiatry 1999, 7, 125–143. [Google Scholar] [CrossRef]
- Gomperts, S.N.; Rao, A.; Craig, A.M.; Malenka, R.C.; Nicoll, R.A. Postsynaptically Silent Synapses in Single Neuron Cultures. Neuron 1998, 21, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panizzutti, R.; Rausch, M.; Zurbrügg, S.; Baumann, D.; Beckmann, N.; Rudin, M. The Pharmacological Stimulation of NMDA Receptors via Co-Agonist Site: An FMRI Study in the Rat Brain. Neurosci. Lett. 2005, 380, 111–115. [Google Scholar] [CrossRef]
- Krystal, J.H. Neuroplasticity as a Target for the Pharmacotherapy of Psychiatric Disorders: New Opportunities for Synergy with Psychotherapy. Biol. Psychiatry 2007, 62, 833–834. [Google Scholar] [CrossRef] [PubMed]
- Guastella, A.J.; Lovibond, P.F.; Dadds, M.R.; Mitchell, P.; Richardson, R. A Randomized Controlled Trial of the Effect of D-Cycloserine on Extinction and Fear Conditioning in Humans. Behav. Res. Ther. 2007, 45, 663–672. [Google Scholar] [CrossRef]
- Klumpers, F.; Denys, D.; Kenemans, J.L.; Grillon, C.; van der Aart, J.; Baas, J.M.P. Testing the Effects of Δ9-THC and D-Cycloserine on Extinction of Conditioned Fear in Humans. J. Psychopharmacol. 2012, 26, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdorf, T.B.; Haaker, J.; Kalisch, R. Long-Term Expression of Human Contextual Fear and Extinction Memories Involves Amygdala, Hippocampus and Ventromedial Prefrontal Cortex: A Reinstatement Study in Two Independent Samples. Soc. Cogn. Affect. Neurosci. 2014, 9, 1973–1983. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, S.G.; Meuret, A.E.; Smits, J.A.J.; Simon, N.M.; Pollack, M.H.; Eisenmenger, K.; Shiekh, M.; Otto, M.W. Augmentation of Exposure Therapy with D-Cycloserine for Social Anxiety Disorder. Arch. Gen. Psychiatry 2006, 63, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Smits, J.A.J.; Rosenfield, D.; Otto, M.W.; Marques, L.; Davis, M.L.; Meuret, A.E.; Simon, N.M.; Pollack, M.H.; Hofmann, S.G. D-Cycloserine Enhancement of Exposure Therapy for Social Anxiety Disorder Depends on the Success of Exposure Sessions. J. Psychiatr. Res. 2013, 47, 1455–1461. [Google Scholar] [CrossRef] [Green Version]
- Kushner, M.G.; Kim, S.W.; Donahue, C.; Thuras, P.; Adson, D.; Kotlyar, M.; McCabe, J.; Peterson, J.; Foa, E.B. D-Cycloserine Augmented Exposure Therapy for Obsessive-Compulsive Disorder. Biol. Psychiatry 2007, 62, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Amaral, O.B.; Roesler, R. Targeting the NMDA Receptor for Fear-Related Disorders. Recent Pat. CNS Drug Discov. 2008, 3, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Ledgerwood, L.; Richardson, R.; Cranney, J. D-Cycloserine and the Facilitation of Extinction of Conditioned Fear: Consequences for Reinstatement. Behav. Neurosci. 2004, 118, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrholm, S.D.; Jovanovic, T.; Vervliet, B.; Myers, K.M.; Davis, M.; Rothbaum, B.O.; Duncan, E.J. Conditioned Fear Extinction and Reinstatement in a Human Fear-Potentiated Startle Paradigm. Learn. Mem. 2006, 13, 681–685. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.L.C.; Milton, A.L.; Everitt, B.J. Reconsolidation and Extinction of Conditioned Fear: Inhibition and Potentiation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 10051–10056. [Google Scholar] [CrossRef] [Green Version]
- Myers, K.M.; Ressler, K.J.; Davis, M. Different Mechanisms of Fear Extinction Dependent on Length of Time since Fear Acquisition. Learn. Mem. 2006, 13, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, K.; Honma, M.; Soshi, T.; Fujii, T.; Kim, Y. Effect of D-Cycloserine and Valproic Acid on the Extinction of Reinstated Fear-Conditioned Responses and Habituation of Fear Conditioning in Healthy Humans: A Randomized Controlled Trial. Psychopharmacology 2011, 218, 589–597. [Google Scholar] [CrossRef]
- Göttlicher, M. Valproic Acid: An Old Drug Newly Discovered as Inhibitor of Histone Deacetylases. Ann. Hematol. 2004, 83 (Suppl. S1), S91–S92. [Google Scholar] [CrossRef]
- Jeon, G.S.; Park, S.-H.; Lee, K.-J.; Lee, M.-S.; Chun, B.-G.; Shin, K.-H. Valproate Prevents MK801-Induced Changes in Brain-Derived Neurotrophic Factor MRNA in the Rat Brain. Eur. J. Pharmacol. 2006, 545, 142–146. [Google Scholar] [CrossRef]
- Miyagi, J.; Oshibuchi, H.; Kasai, A.; Inada, K.; Ishigooka, J. Valproic Acid Inhibits Excess Dopamine Release in Response to a Fear-Conditioned Stimulus in the Basolateral Complex of the Amygdala of Methamphetamine-Sensitized Rats. Eur. J. Pharmacol. 2014, 730, 20–25. [Google Scholar] [CrossRef]
- Schiller, D.; Monfils, M.-H.; Raio, C.M.; Johnson, D.C.; LeDoux, J.E.; Phelps, E.A. Preventing the Return of Fear in Humans Using Reconsolidation Update Mechanisms. Nature 2010, 463, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyama, K.; Honma, M.; Yoshiike, T.; Kim, Y. Valproic Acid but Not D-Cycloserine Facilitates Sleep-Dependent Offline Learning of Extinction and Habituation of Conditioned Fear in Humans. Neuropharmacology 2013, 64, 424–431. [Google Scholar] [CrossRef]
- Ebrahimi, C.; Gechter, J.; Lueken, U.; Schlagenhauf, F.; Wittchen, H.U.; Hamm, A.O.; Ströhle, A. Augmenting Extinction Learning with D-Cycloserine Reduces Return of Fear: A Randomized, Placebo-Controlled FMRI Study. Neuropsychopharmacology 2020, 45, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Malan-Müller, S.; de Souza, V.B.C.; Daniels, W.M.U.; Seedat, S.; Robinson, M.D.; Hemmings, S.M.J. Shedding Light on the Transcriptomic Dark Matter in Biological Psychiatry: Role of Long Noncoding RNAs in D-Cycloserine-Induced Fear Extinction in Posttraumatic Stress Disorder. OMICS 2020, 24, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Malan-Müller, S.; Fairbairn, L.; Hart, S.; Daniels, W.M.U.; Jalali Sefid Dashti, M.; Kidd, M.; Seedat, S.; Gamieldien, J.; Hemmings, S.M.J. The Role of MicroRNAs in the Therapeutic Action of D-Cycloserine in a Post-Traumatic Stress Disorder Animal Model: An Exploratory Study. Psychiatr. Genet. 2017, 27, 139–151. [Google Scholar] [CrossRef]
- Inslicht, S.S.; Niles, A.N.; Metzler, T.J.; Lipshitz, S.L.; Otte, C.; Milad, M.R.; Orr, S.P.; Marmar, C.R.; Neylan, T.C. Randomized Controlled Experimental Study of Hydrocortisone and D-Cycloserine Effects on Fear Extinction in PTSD. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2022, 47, 1945–1952. [Google Scholar] [CrossRef]
- Malvaez, M.; Sanchis-Segura, C.; Vo, D.; Lattal, K.M.; Wood, M.A. Modulation of Chromatin Modification Facilitates Extinction of Cocaine-Induced Conditioned Place Preference. Biol. Psychiatry 2010, 67, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.J.; Karra, A.S.; Monteggia, L.M. Histone Deacetylases Govern Cellular Mechanisms Underlying Behavioral and Synaptic Plasticity in the Developing and Adult Brain. Behav. Pharmacol. 2010, 21, 409–419. [Google Scholar] [CrossRef]
- Roozendaal, B.; Castello, N.A.; Vedana, G.; Barsegyan, A.; McGaugh, J.L. Noradrenergic Activation of the Basolateral Amygdala Modulates Consolidation of Object Recognition Memory. Neurobiol. Learn. Mem. 2008, 90, 576–579. [Google Scholar] [CrossRef] [Green Version]
- Joëls, M.; Fernandez, G.; Roozendaal, B. Stress and Emotional Memory: A Matter of Timing. Trends Cogn. Sci. 2011, 15, 280–288. [Google Scholar] [CrossRef]
- Barsegyan, A.; McGaugh, J.L.; Roozendaal, B. Noradrenergic Activation of the Basolateral Amygdala Modulates the Consolidation of Object-in-Context Recognition Memory. Front. Behav. Neurosci. 2014, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- McGaugh, J.L. Making Lasting Memories: Remembering the Significant. Proc. Natl. Acad. Sci. USA 2013, 110 (Suppl. 2), 10402–10407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atucha, E.; Vukojevic, V.; Fornari, R.V.; Ronzoni, G.; Demougin, P.; Peter, F.; Atsak, P.; Coolen, M.W.; Papassotiropoulos, A.; McGaugh, J.L.; et al. Noradrenergic Activation of the Basolateral Amygdala Maintains Hippocampus-Dependent Accuracy of Remote Memory. Proc. Natl. Acad. Sci. USA 2017, 114, 9176–9181. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.I.; LaLumiere, R.T.; Buen, T.V.; Bermudez-Rattoni, F.; McGaugh, J.L. Blockade of Noradrenergic Receptors in the Basolateral Amygdala Impairs Taste Memory. Eur. J. Neurosci. 2003, 18, 2605–2610. [Google Scholar] [CrossRef]
- LaLumiere, R.T.; Buen, T.-V.; McGaugh, J.L. Post-Training Intra-Basolateral Amygdala Infusions of Norepinephrine Enhance Consolidation of Memory for Contextual Fear Conditioning. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 6754–6758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeter, M.; Kindt, M. Noradrenergic Enhancement of Associative Fear Memory in Humans. Neurobiol. Learn. Mem. 2011, 96, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Soeter, M.; Kindt, M. Disrupting Reconsolidation: Pharmacological and Behavioral Manipulations. Learn. Mem. 2011, 18, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Soeter, M.; Kindt, M. Stimulation of the Noradrenergic System during Memory Formation Impairs Extinction Learning but Not the Disruption of Reconsolidation. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2012, 37, 1204–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdorf, T.B.; Haaker, J.; Fadai, T.; Kalisch, R. No Evidence for Enhanced Extinction Memory Consolidation through Noradrenergic Reuptake Inhibition—Delayed Memory Test and Reinstatement in Human FMRI. Psychopharmacology 2014, 231, 1949–1962. [Google Scholar] [CrossRef]
- Kausche, F.M.; Zerbes, G.; Kampermann, L.; Müller, J.C.; Wiedemann, K.; Büchel, C.; Schwabe, L. Noradrenergic Stimulation Increases Fear Memory Expression. Eur. Neuropsychopharmacol. 2021, 43, 71–81. [Google Scholar] [CrossRef]
- Bambico, F.R.; Katz, N.; Debonnel, G.; Gobbi, G. Cannabinoids Elicit Antidepressant-like Behavior and Activate Serotonergic Neurons through the Medial Prefrontal Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 11700–11711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbi, G.; Bambico, F.R.; Mangieri, R.; Bortolato, M.; Campolongo, P.; Solinas, M.; Cassano, T.; Morgese, M.G.; Debonnel, G.; Duranti, A.; et al. Antidepressant-like Activity and Modulation of Brain Monoaminergic Transmission by Blockade of Anandamide Hydrolysis. Proc. Natl. Acad. Sci. USA 2005, 102, 18620–18625. [Google Scholar] [CrossRef] [Green Version]
- Bluett, R.J.; Báldi, R.; Haymer, A.; Gaulden, A.D.; Hartley, N.D.; Parrish, W.P.; Baechle, J.; Marcus, D.J.; Mardam-Bey, R.; Shonesy, B.C.; et al. Endocannabinoid Signalling Modulates Susceptibility to Traumatic Stress Exposure. Nat. Commun. 2017, 8, 14782. [Google Scholar] [CrossRef] [Green Version]
- Van Bockstaele, E.J. Cannabinoid Receptor Signaling and Modulation of Monoamines: Implications for Psychiatric and Neurological Disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 38, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Hill, T.D.M.; Cascio, M.-G.; Romano, B.; Duncan, M.; Pertwee, R.G.; Williams, C.M.; Whalley, B.J.; Hill, A.J. Cannabidivarin-Rich Cannabis Extracts Are Anticonvulsant in Mouse and Rat via a CB1 Receptor-Independent Mechanism. Br. J. Pharmacol. 2013, 170, 679–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabik, N.L.; Iadipaolo, A.S.; Marusak, H.A.; Peters, C.; Burghardt, K.; Rabinak, C.A. A Common Genetic Variant in Fatty Acid Amide Hydrolase Is Linked to Alterations in Fear Extinction Neural Circuitry in a Racially Diverse, Nonclinical Sample of Adults. J. Neurosci. Res. 2022, 100, 744–761. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.M.; Asratian, A.; Lindé, J.; Holm, L.; Nätt, D.; Augier, G.; Stensson, N.; Vecchiarelli, H.A.; Balsevich, G.; Aukema, R.J.; et al. Protective Effects of Elevated Anandamide on Stress and Fear-Related Behaviors: Translational Evidence from Humans and Mice. Mol. Psychiatry 2020, 25, 993–1005. [Google Scholar] [CrossRef]
- Rafiei, D.; Kolla, N.J. Elevated Brain Fatty Acid Amide Hydrolase Induces Depressive-Like Phenotypes in Rodent Models: A Review. Int. J. Mol. Sci. 2021, 22, 1047. [Google Scholar] [CrossRef]
- Mayo, L.M.; Asratian, A.; Lindé, J.; Morena, M.; Haataja, R.; Hammar, V.; Augier, G.; Hill, M.N.; Heilig, M. Elevated Anandamide, Enhanced Recall of Fear Extinction, and Attenuated Stress Responses Following Inhibition of Fatty Acid Amide Hydrolase: A Randomized, Controlled Experimental Medicine Trial. Biol. Psychiatry 2020, 87, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Gunduz-Cinar, O.; MacPherson, K.P.; Cinar, R.; Gamble-George, J.; Sugden, K.; Williams, B.; Godlewski, G.; Ramikie, T.S.; Gorka, A.X.; Alapafuja, S.O.; et al. Convergent Translational Evidence of a Role for Anandamide in Amygdala-Mediated Fear Extinction, Threat Processing and Stress-Reactivity. Mol. Psychiatry 2013, 18, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.M.; Okorokov, A.L.; Hill, M.N.; Bras, J.T.; Lee, M.-C.; Li, S.; Gossage, S.J.; van Drimmelen, M.; Morena, M.; Houlden, H.; et al. Microdeletion in a FAAH Pseudogene Identified in a Patient with High Anandamide Concentrations and Pain Insensitivity. Br. J. Anaesth. 2019, 123, e249–e253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, M.P.; Stein, M.B.; Simmons, A.N.; Risbrough, V.B.; Halter, R.; Chaplan, S.R. The Effects of FAAH Inhibition on the Neural Basis of Anxiety-Related Processing in Healthy Male Subjects: A Randomized Clinical Trial. Neuropsychopharmacology 2021, 46, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.M.; Rabinak, C.A.; Hill, M.N.; Heilig, M. Targeting the Endocannabinoid System in the Treatment of Posttraumatic Stress Disorder: A Promising Case of Preclinical-Clinical Translation? Biol. Psychiatry 2022, 91, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Ney, L.J.; Matthews, A.; Hsu, C.-M.K.; Zuj, D.V.; Nicholson, E.; Steward, T.; Nichols, D.; Graham, B.; Harrison, B.; Bruno, R.; et al. Cannabinoid Polymorphisms Interact with Plasma Endocannabinoid Levels to Predict Fear Extinction Learning. Depress. Anxiety 2021, 38, 1087–1099. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Vitale, F.; Battaglia, S.; Avenanti, A. Early Right Motor Cortex Response to Happy and Fearful Facial Expressions: A TMS Motor-Evoked Potential Study. Brain Sci. 2021, 11, 1203. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Vitale, F.; Avenanti, A. Behavioral Inhibition System Sensitivity Enhances Motor Cortex Suppression When Watching Fearful Body Expressions. Brain Struct. Funct. 2017, 222, 3267–3282. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Vitale, F.; Avenanti, A. Early Changes in Corticospinal Excitability When Seeing Fearful Body Expressions. Sci. Rep. 2015, 5, 14122. [Google Scholar] [CrossRef] [Green Version]
- Bertini, C.; Làdavas, E. Fear-Related Signals Are Prioritised in Visual, Somatosensory and Spatial Systems. Neuropsychologia 2021, 150, 107698. [Google Scholar] [CrossRef]
- Tamietto, M.; de Gelder, B. Neural Bases of the Non-Conscious Perception of Emotional Signals. Nat. Rev. Neurosci. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Kuhn, M.; Gerlicher, A.M.V.; Lonsdorf, T.B. Navigating the manyverse of skin conductance response quantification approaches—A direct comparison of trough-to-peak, baseline correction, and model-based approaches in Ledalab and PsPM. Psychophysiology 2022, 59, e14058. [Google Scholar] [CrossRef]
- Lonsdorf, T.B.; Klingelhöfer-Jens, M.; Andreatta, M.; Beckers, T.; Chalkia, A.; Gerlicher, A.; Jentsch, V.L.; Meir Drexler, S.; Mertens, G.; Richter, J.; et al. Navigating the Garden of Forking Paths for Data Exclusions in Fear Conditioning Research. Elife 2019, 8, e52465. [Google Scholar] [CrossRef] [PubMed]
- Schiller, D.; Levy, I.; Niv, Y.; LeDoux, J.E.; Phelps, E.A. From Fear to Safety and Back: Reversal of Fear in the Human Brain. J. Neurosci. 2008, 28, 11517–11525. [Google Scholar] [CrossRef] [Green Version]
- Schiller, D.; Delgado, M.R. Overlapping Neural Systems Mediating Extinction, Reversal and Regulation of Fear. Trends Cogn. Sci. 2010, 14, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, P.J.; Seemann, J.R.; Maren, S. Can Fear Extinction Be Enhanced? A Review of Pharmacological and Behavioral Findings. Brain Res. Bull. 2014, 105, 46–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirru, M.; Véronneau-Veilleux, F.; Nekka, F.; Ursino, M. Phasic Dopamine Changes and Hebbian Mechanisms during Probabilistic Reversal Learning in Striatal Circuits: A Computational Study. Int. J. Mol. Sci. 2022, 23, 3452. [Google Scholar] [CrossRef] [PubMed]
- Milad, M.R.; Rosenbaum, B.L.; Simon, N.M. Neuroscience of Fear Extinction: Implications for Assessment and Treatment of Fear-Based and Anxiety Related Disorders. Behav. Res. Ther. 2014, 62, 17–23. [Google Scholar] [CrossRef]
- Milad, M.R.R.; Quirk, G.J.J. Neurons in Medial Prefrontal Cortex Signal Memory for Fear Extinction. Nature 2002, 420, 70–74. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, I.; Temprano-Carazo, S.; Jeremic, D.; Delgado-Garcia, J.M.; Gruart, A.; Navarro-López, J.D.; Jiménez-Díaz, L. Recognition Memory Induces Natural LTP-like Hippocampal Synaptic Excitation and Inhibition. Int. J. Mol. Sci. 2022, 23, 806. [Google Scholar] [CrossRef]
- Luchkina, N.V.; Bolshakov, V.Y. Mechanisms of Fear Learning and Extinction: Synaptic Plasticity-Fear Memory Connection. Psychopharmacology 2019, 236, 163–182. [Google Scholar] [CrossRef]
- Mahan, A.L.; Ressler, K.J. Fear Conditioning, Synaptic Plasticity and the Amygdala: Implications for Posttraumatic Stress Disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Romei, V.; Chiappini, E.; Hibbard, P.B.; Avenanti, A. Empowering Reentrant Projections from V5 to V1 Boosts Sensitivity to Motion. Curr. Biol. 2016, 26, 2155–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappini, E.; Silvanto, J.; Hibbard, P.B.; Avenanti, A.; Romei, V. Strengthening Functionally Specific Neural Pathways with Transcranial Brain Stimulation. Current Biol. CB 2018, 28, R735–R736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, F.; Chiappini, E.; Avenanti, A. Enhanced Action Performance Following TMS Manipulation of Associative Plasticity in Ventral Premotor-Motor Pathway. Neuroimage 2018, 183, 847–858. [Google Scholar] [CrossRef]
- Turrini, S.; Fiori, F.; Chiappini, E.; Santarnecchi, E.; Romei, V.; Avenanti, A. Gradual Enhancement of Corticomotor Excitability during Cortico-Cortical Paired Associative Stimulation. Sci. Rep. 2022, 12, 14670. [Google Scholar] [CrossRef] [PubMed]
- Turrini, S.; Bevacqua, N.; Cataneo, A.; Chiappini, E.; Fiori, F.; Candidi, M.; Avenanti, A. Transcranial Cortico-Cortical Paired Associative Stimulation (CcPAS) over Ventral Premotor-Motor Pathways Enhances Action Performance and Corticomotor Excitability in Young Adults More than in Elderly Adults. Front. Aging Neurosci. 2023, 15, 1119508. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.; Kim, J.; Lee, J.; Park, S.; Song, B.; Kim, J.; An, B.; Park, K.; Lee, H.W.; Lee, S.; et al. Reversible Plasticity of Fear Memory-Encoding Amygdala Synaptic Circuits Even after Fear Memory Consolidation. PLoS ONE 2011, 6, e24260. [Google Scholar] [CrossRef] [PubMed]
- Singewald, N.; Schmuckermair, C.; Whittle, N.; Holmes, A.; Ressler, K.J. Pharmacology of Cognitive Enhancers for Exposure-Based Therapy of Fear, Anxiety and Trauma-Related Disorders. Pharmacol. Ther. 2015, 149, 150–190. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Török, N.; Vécsei, L. Are 5-HT(1) Receptor Agonists Effective Anti-Migraine Drugs? Expert Opin. Pharmacother. 2021, 22, 1221–1225. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Vécsei, L. Integrating Armchair, Bench, and Bedside Research for Behavioral Neurology and Neuropsychiatry: Editorial. Biomedicines 2022, 10, 2999. [Google Scholar] [CrossRef]
- Tanaka, M.; Spekker, E.; Szabó, Á.; Polyák, H.; Vécsei, L. Modelling the Neurodevelopmental Pathogenesis in Neuropsychiatric Disorders. Bioactive Kynurenines and Their Analogues as Neuroprotective Agents-in Celebration of 80th Birthday of Professor Peter Riederer. J. Neural Transm. 2022, 129, 627–642. [Google Scholar] [CrossRef]
- Balogh, L.; Tanaka, M.; Török, N.; Taguchi, S. Crosstalk between Existential Phenomenological Psychotherapy and Neurological Sciences in Mood and Anxiety Disorders. Biomedicines 2021, 9, 340. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue “Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry”. Biomedicines 2021, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Monitoring the Kynurenine System: Concentrations, Ratios or What Else? Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2021, 30, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Schally, A.V.; Telegdy, G. Neurotransmission of the Antidepressant-like Effects of the Growth Hormone-Releasing Hormone Antagonist MZ-4-71. Behav. Brain Res. 2012, 228, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Fanni, T.; Poly, H.; Szab, Á.; Yvette, M. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 743. [Google Scholar] [CrossRef]
- Telegdy, G.; Tanaka, M.; Schally, A.V. Effects of the Growth Hormone-Releasing Hormone (GH-RH) Antagonist on Brain Functions in Mice. Behav. Brain Res. 2011, 224, 155–158. [Google Scholar] [CrossRef]
- Tanaka, M.; Kádár, K.; Tóth, G.; Telegdy, G. Antidepressant-like Effects of Urocortin 3 Fragments. Brain Res. Bull. 2011, 84, 414–418. [Google Scholar] [CrossRef]
- Phelps, E.A.; Delgado, M.R.; Nearing, K.I.; Ledoux, J.E. Extinction Learning in Humans: Role of the Amygdala and VmPFC. Neuron 2004, 43, 897–905. [Google Scholar] [CrossRef] [Green Version]
- LeDoux, J.E. Emotion Circuits in the Brain. Annu. Rev. Neurosci. 2000, 23, 155–184. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan-Kynurenine Metabolic System. Cells 2022, 11, 2607. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue “Dissecting Neurological and Neuropsychiatric Diseases: Neurodegeneration and Neuroprotection”. Int. J. Mol. Sci. 2022, 23, 6991. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G. The Mechanisms of Action of Valproate in Neuropsychiatric Disorders: Can We See the Forest for the Trees? Cell. Mol. Life Sci. 2007, 64, 2090–2103. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.B.; McLaughlin, L.D.; Ebenezer, P.J.; Nair, A.R.; Francis, J. Valproic Acid Effects in the Hippocampus and Prefrontal Cortex in an Animal Model of Post-Traumatic Stress Disorder. Behav. Brain Res. 2014, 268, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Roozendaal, B.; Hui, G.K.; Hui, I.R.; Berlau, D.J.; McGaugh, J.L.; Weinberger, N.M. Basolateral Amygdala Noradrenergic Activity Mediates Corticosterone-Induced Enhancement of Auditory Fear Conditioning. Neurobiol. Learn. Mem. 2006, 86, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Berlau, D.J.; McGaugh, J.L. Enhancement of Extinction Memory Consolidation: The Role of the Noradrenergic and GABAergic Systems within the Basolateral Amygdala. Neurobiol. Learn. Mem. 2006, 86, 123–132. [Google Scholar] [CrossRef]
- Sehlmeyer, C.; Schöning, S.; Zwitserlood, P.; Pfleiderer, B.; Kircher, T.; Arolt, V.; Konrad, C. Human Fear Conditioning and Extinction in Neuroimaging: A Systematic Review. PLoS ONE 2009, 4, e5865. [Google Scholar] [CrossRef] [Green Version]
- Milad, M.R.; Wright, C.I.; Orr, S.P.; Pitman, R.K.; Quirk, G.J.; Rauch, S.L. Recall of Fear Extinction in Humans Activates the Ventromedial Prefrontal Cortex and Hippocampus in Concert. Biol. Psychiatry 2007, 62, 446–454. [Google Scholar] [CrossRef]
- Bluett, R.J.; Gamble-George, J.C.; Hermanson, D.J.; Hartley, N.D.; Marnett, L.J.; Patel, S. Central Anandamide Deficiency Predicts Stress-Induced Anxiety: Behavioral Reversal through Endocannabinoid Augmentation. Transl. Psychiatry 2014, 4, e408. [Google Scholar] [CrossRef] [Green Version]
- Bortolato, M.; Mangieri, R.A.; Fu, J.; Kim, J.H.; Arguello, O.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Antidepressant-like Activity of the Fatty Acid Amide Hydrolase Inhibitor URB597 in a Rat Model of Chronic Mild Stress. Biol. Psychiatry 2007, 62, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.M.; Vecchiarelli, H.A.; Morena, M.; Lee, T.T.Y.; Hermanson, D.J.; Kim, A.B.; McLaughlin, R.J.; Hassan, K.I.; Kühne, C.; Wotjak, C.T.; et al. Corticotropin-Releasing Hormone Drives Anandamide Hydrolysis in the Amygdala to Promote Anxiety. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 3879–3892. [Google Scholar] [CrossRef] [Green Version]
- Morena, M.; Patel, S.; Bains, J.S.; Hill, M.N. Neurobiological Interactions Between Stress and the Endocannabinoid System. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 80–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; Campolongo, P.; Yehuda, R.; Patel, S. Integrating Endocannabinoid Signaling and Cannabinoids into the Biology and Treatment of Posttraumatic Stress Disorder. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 80–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.N.; McLaughlin, R.J.; Morrish, A.C.; Viau, V.; Floresco, S.B.; Hillard, C.J.; Gorzalka, B.B. Suppression of Amygdalar Endocannabinoid Signaling by Stress Contributes to Activation of the Hypothalamic-Pituitary-Adrenal Axis. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2009, 34, 2733–2745. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, A.; Dörfel, D.; Diers, K.; Witt, S.H.; Strobel, A.; Brocke, B. Impact of FAAH Genetic Variation on Fronto-Amygdala Function during Emotional Processing. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Bandelow, B.; Michaelis, S. Epidemiology of Anxiety Disorders in the 21st Century. Dialogues Clin. Neurosci. 2015, 17, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Black, D.W. Efficacy of Combined Pharmacotherapy and Psychotherapy versus Monotherapy in the Treatment of Anxiety Disorders. CNS Spectr. 2006, 11 (Suppl. 12), 29–33. [Google Scholar] [CrossRef]
- Bitencourt, R.M.; Takahashi, R.N. Cannabidiol as a Therapeutic Alternative for Post-Traumatic Stress Disorder: From Bench Research to Confirmation in Human Trials. Front. Neurosci. 2018, 12, 502. [Google Scholar] [CrossRef] [Green Version]
- Kelmendi, B.; Adams, T.G.; Southwick, S.; Abdallah, C.G.; Krystal, J.H. Posttraumatic Stress Disorder: An Integrated Overview and Neurobiological Rationale for Pharmacology. Clin. Psychol. Publ. Div. Clin. Psychol. Am. Psychol. Assoc. 2017, 24, 281–297. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Averill, L.A.; Akiki, T.J.; Raza, M.; Averill, C.L.; Gomaa, H.; Adikey, A.; Krystal, J.H. The Neurobiology and Pharmacotherapy of Posttraumatic Stress Disorder. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 171–189. [Google Scholar] [CrossRef]
- Lefaucheur, J.-P.; Aleman, A.; Baeken, C.; Benninger, D.H.; Brunelin, J.; Di Lazzaro, V.; Filipović, S.R.; Grefkes, C.; Hasan, A.; Hummel, F.C.; et al. Evidence-Based Guidelines on the Therapeutic Use of Repetitive Transcranial Magnetic Stimulation (RTMS): An Update (2014–2018). Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 474–528. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Hegefeld, T.L.; Petersen, M.; Patterson, J.C., 2nd; Yossi, C.; Slizewski, J.; Osumi, A.; Cornett, E.M.; Kaye, A.; Kaye, J.S.; et al. Transcranial Magnetic Stimulation for Post-Traumatic Stress Disorder. Front. Psychiatry 2022, 13, 701348. [Google Scholar] [CrossRef] [PubMed]
- Guhn, A.; Dresler, T.; Andreatta, M.; Müller, L.D.; Hahn, T.; Tupak, S.V.; Polak, T.; Deckert, J.; Herrmann, M.J. Medial Prefrontal Cortex Stimulation Modulates the Processing of Conditioned Fear. Front. Behav. Neurosci. 2014, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asthana, M.; Nueckel, K.; Mühlberger, A.; Neueder, D.; Polak, T.; Domschke, K.; Deckert, J.; Herrmann, M.J. Effects of Transcranial Direct Current Stimulation on Consolidation of Fear Memory. Front. Psychiatry 2013, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Study | Group (N) | Pharmacological Treatment | Mechanism of Action | Experimental Paradigm | Phase of Fear Learning | CSs | US | Psychophysiological Measure | Main Findings |
|---|---|---|---|---|---|---|---|---|---|
| Kuriyama et al. [111] | DCS treatment (16) VPA treatment (14) Combined VPA–DCS treatment (15) Placebo treatment (15) | 100 mg of powdered DCS 400 mg of granulated VPA 1200 mg of lactose (placebo) | DCS is an agonist of the NMDA receptor VPA is an inhibitor of GABA transaminase and HDAC | 3 days | Acquisition Extinction Recall Reinstatement | Geometric figures | Electric shock | SCR | DCS and VPA enhanced extinction learning of the fear CRs in the post-recall reinstatement phase, but not in the extinction recall phase |
| Kuriyama et al. [116] | DCS × Sleep treatment (14) DCS × Wake treatment (14) VPA × Sleep treatment (15) VPA × Wake treatment (15) Placebo × Sleep treatment (15) Placebo × Wake treatment (14) | 100 mg of powdered DCS 400 mg of granulated VPA 1000 mg of lactose (placebo) | DCS is an agonist of the NMDA receptor VPA is an inhibitor of GABA transaminase and HDAC | 1 day | Acquisition Extinction Recall Reinstatement | Geometric figures | Electric shock | SCR | VPA treatment reduced fear CRs in the extinction and acquisition phases during the second learning session DCS treatment blocked the effect of the reinforced CS–US pairing only in the waking group; VPA blocked the effect of the reinforced CS–US pairing only in the sleep group |
| Ebrahimi et al. [116] | DCS treatment group (17) Placebo treatment (20) | 50 mg of powdered DCS Placebo | DCS is an agonist of the NMDA receptor | 3 days | Habituation Acquisition Extinction Recall | Ekman faces | Auditory tone | SCR and fMRI | DCS administration enhanced extinction memory retention by preventing differential CRs from extinction learning to recall in subjective arousal ratings and attenuating BOLD responses in the hippocampus and amygdala |
| Study | Group (N) | Pharmacological Treatment | Mechanism of Action | Experimental Paradigm | Phase of Fear Learning | CSs | US | Psychophysiological Measure | Main Findings |
|---|---|---|---|---|---|---|---|---|---|
| Lonsdorf et al. [133] | RBX treatment (23) Placebo treatment (19) | 4 mg of RBX Placebo | Reboxetine is an inhibitor of noradrenaline reuptake | 3 days | Acquisition Extinction | Symbols shown in three different contexts | Electrotactile shock | SCR and fMRI | No SCR differences between groups at the behavioral level Before reinstatement, only the placebo group showed higher activation in vmPFC for CS cues After reinstatement, the RBX group showed higher amygdala activation for CS cues |
| Kausche et al. [134] | Placebo treatment (31) CORT treatment (31) YOH treatment (34) CORT+YOH treatment (29) | 20 mg of cortisol 20 mg of YOH 20 mg of cortisol and YOH Placebo | Cortisol is an agonist of glucocorticoid receptor and annexin A1 Yohimbine is an α2-adrenergic blocking agent | 2 days | Habituation Acquisition | Eight neutral faces | Electric shock | SCR | The YOH group showed higher SCR across a similarity continuum from CS+ to CS− (increased responses to the CS+) Cortisol did not enhance fear memory expression, but increased fear generalization |
| Soeter and Kindt [132] | Yohimbine treatment (20) Propranolol treatment (20) | 40 mg of YOH 20 mg of propranolol Placebo | Yohimbine is an α2-adrenergic blocking agent | 3 days | Acquisition Recall Extinction | Three images | Auditory tone | SCR Systolic and diastolic blood pressure Amylase level | The YOH group showed higher startle fear responses during fear memory reactivation on day 2 The propanolol group showed a reduction in startle fear responses during reconsolidation and extinction |
| Study | Group (N) | Pharmacological Treatment | Mechanism of Action | Experimental Paradigm | Phase of fear Learning | CSs | US | Psychophysiological Measure | Main Findings |
|---|---|---|---|---|---|---|---|---|---|
| Mayo et al. [143] | FAAH inhibitor treatment (16) Placebo treatment (29) | 4 mg/day of PF-04457845 (FAAH inhibitor) Placebo | PF-04457845 is an inhibitor of FAAH | 10 days | Habituation Acquisition Extinction Recall Renewal | Two lamps shown in two different contexts | Auditory tone | SCR, ECG, and EMG | The FAAH inhibitor group showed lower responses to the CS+ on day 2, indicating enhanced recall of extinction memory |
| Paulus et al. [146] | FAAH inhibitor treatment (22) Placebo treatment (21) | 100 mg/day of JNJ-42165279 (FAAH inhibitor) Placebo | JNJ-42165279 is an inhibitor of FAAH | 4 days | Habituation Acquisition Extinction | Fractal stimuli | Auditory tone | fMRI | No differences between groups during the acquisition and extinction phases |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battaglia, S.; Di Fazio, C.; Vicario, C.M.; Avenanti, A. Neuropharmacological Modulation of N-methyl-D-aspartate, Noradrenaline and Endocannabinoid Receptors in Fear Extinction Learning: Synaptic Transmission and Plasticity. Int. J. Mol. Sci. 2023, 24, 5926. https://doi.org/10.3390/ijms24065926
Battaglia S, Di Fazio C, Vicario CM, Avenanti A. Neuropharmacological Modulation of N-methyl-D-aspartate, Noradrenaline and Endocannabinoid Receptors in Fear Extinction Learning: Synaptic Transmission and Plasticity. International Journal of Molecular Sciences. 2023; 24(6):5926. https://doi.org/10.3390/ijms24065926
Chicago/Turabian StyleBattaglia, Simone, Chiara Di Fazio, Carmelo M. Vicario, and Alessio Avenanti. 2023. "Neuropharmacological Modulation of N-methyl-D-aspartate, Noradrenaline and Endocannabinoid Receptors in Fear Extinction Learning: Synaptic Transmission and Plasticity" International Journal of Molecular Sciences 24, no. 6: 5926. https://doi.org/10.3390/ijms24065926