
Genome-Wide Investigation and Co-Expression Network Analysis of SBT Family Gene in Gossypium

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification of SBT Gene Family Members in Cotton
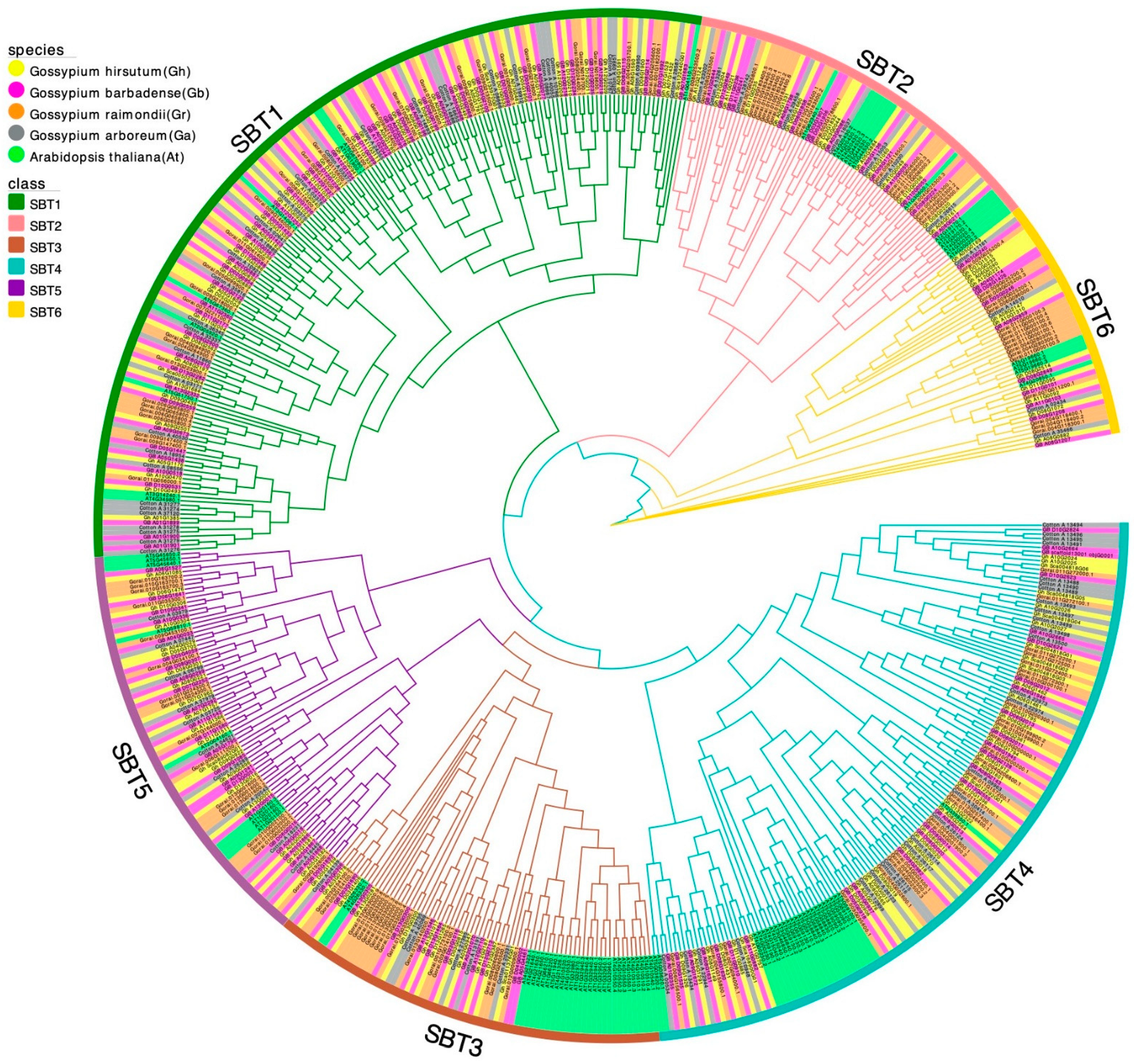
2.2. Phylogenetic Analysis of SBT Gene Family Members in Cotton
2.3. Gene Structure Analysis of SBT Gene Family Members in Cotton
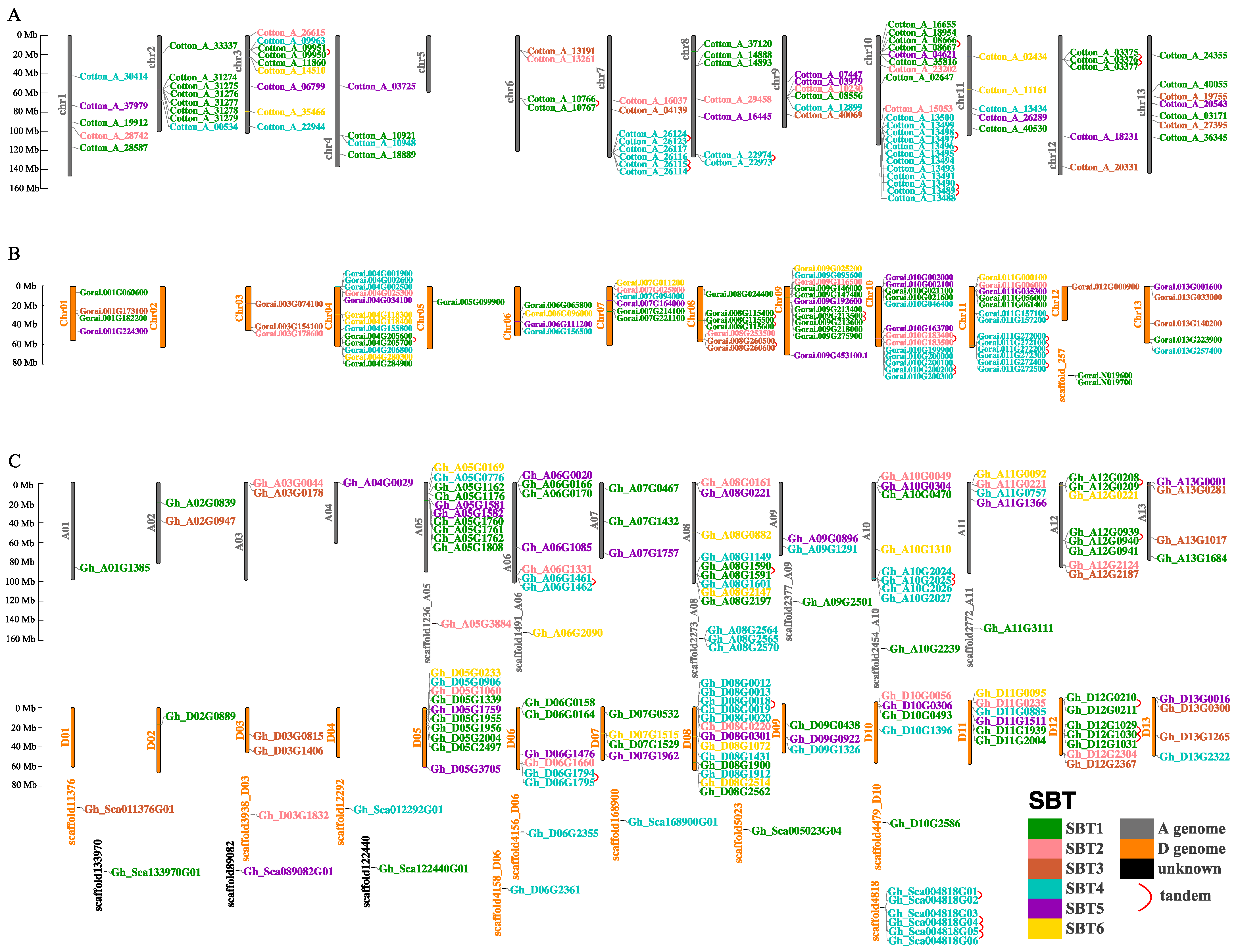
2.4. Chromosomal Localization, Collinearity Analysis and Selective Pressure Analysis of the SBT Family Genes in Cotton
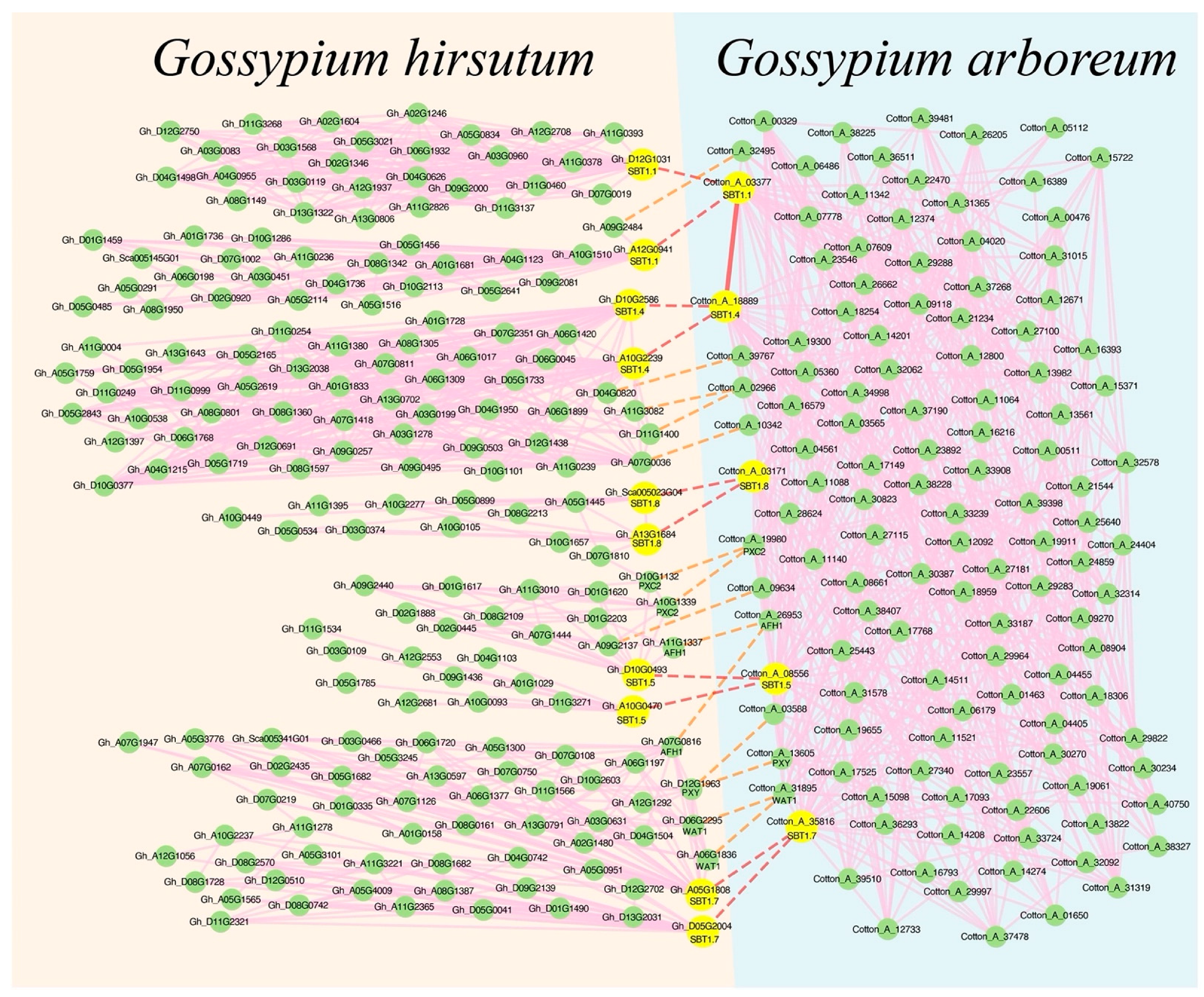
2.5. Co-expression Network Analysis of Gossypium arboreum SBT
2.6. Comparison of Co-Expression Networks between Arabidopsis thaliana and Different Cotton Species
3. Discussion
3.1. Evolution of Cotton SBT Family Genes
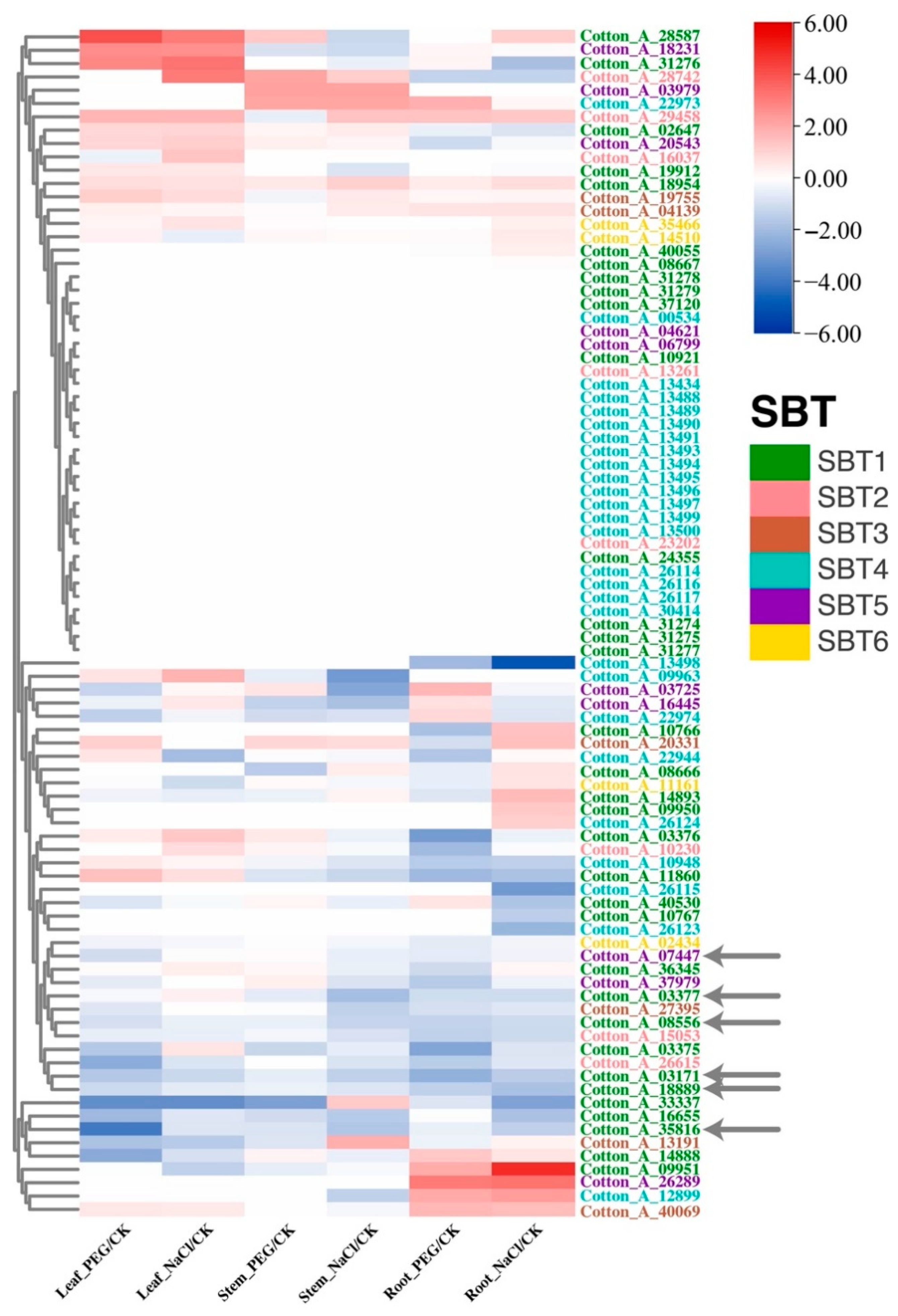
3.2. Cotton SBT Gene Function during Environmental Response
4. Materials and Methods
4.1. Identification of SBT Gene Family Members in Cotton
4.2. Sequence Alignment and Phylogenetic Analysis
4.3. Cotton Gene Structure, Location Display, Cotton Collinearity Analysis, Calculation of Selection Pressure for Duplicated Gene Pairs
4.4. Co-Expression Network Analysis and Functional Enrichment Analysis
4.5. Three-Dimensional Structure Prediction for SBT Protein
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vierstra, R.D. The ubiquitin-26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 2009, 10, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Schaller, A.; Stintzi, A.; Rivas, S.; Serrano, I.; Chichkova, N.V.; Vartapetian, A.B.; Martínez, D.; Guiamét, J.J.; Sueldo, D.J.; van der Hoorn, R.; et al. From structure to function—A family portrait of plant subtilases. New Phytol. 2018, 218, 901–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, A.; Stintzi, A.; Graff, L. Subtilases—Versatile tools for protein turnover, plant development, and interactions with the environment. Physiol. Plant 2012, 145, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.; Wlodawer, A. Catalytic triads and their relatives. Trends Biochem. Sci. 1998, 23, 347–352. [Google Scholar] [CrossRef]
- Jordá, L.; Coego, A.; Conejero, V.; Vera, P. A genomic cluster containing four differentially regulated subtilisin-like processing protease genes is in tomato plants. J. Biol. Chem. 1999, 274, 2360–2365. [Google Scholar] [CrossRef] [Green Version]
- Mahon, P.; Bateman, A. The PA domain: A protease-associated domain. Protein Sci. 2000, 9, 1930–1934. [Google Scholar] [CrossRef] [Green Version]
- Murayama, K.; Kato-Murayama, M.; Hosaka, T.; Sotokawauchi, A.; Yokoyama, S.; Arima, K.; Shirouzu, M. Crystal structure of cucumisin, a subtilisin-like endoprotease from Cucumis melo L. J. Mol. Biol. 2012, 423, 386–396. [Google Scholar] [CrossRef]
- Cedzich, A.; Huttenlocher, F.; Kuhn, B.M.; Pfannstiel, J.; Gabler, L.; Stintzi, A.; Schaller, A. The protease-associated (PA) domain and C-terminal extension are required for zymogen processing, sorting within the secretory pathway, and activity of tomato subtilase 3 (SlSBT3). J. Biol. Chem. 2009, 284, 14068–14078. [Google Scholar] [CrossRef] [Green Version]
- Rautengarten, C.; Steinhauser, D.; Büssis, D.; Stintzi, A.; Schaller, A.; Kopka, J.; Altmann, T. Inferring hypotheses on functional relationships of genes: Analysis of the Arabidopsis thaliana subtilase gene family. PLoS Comput. Biol. 2005, 1, e40. [Google Scholar] [CrossRef] [Green Version]
- Berger, D.; Altmann, T. A subtilisin-like serine protease involved in the regulation of stomatal density and distribution in Arab. Thaliana. Genes Dev. 2000, 14, 1119–1131. [Google Scholar] [CrossRef]
- Von Groll, U.; Berger, D.; Altmann, T. The subtilisin-like serine protease SDD1 mediates cell-to-cell signaling during Arabidopsis stomatal development. Plant Cell 2002, 14, 1527–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Guo, Q.; Guo, Y.; Yang, J.; Wang, M.; Duan, X.; Niu, J.; Liu, S.; Zhang, J.; Lu, Y.; et al. Arabidopsis subtilase SASP is involved in the regulation of ABA signaling and drought tolerance by interacting with OPEN STOMATA 1. J. Exp. Bot. 2018, 69, 4403–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Onouchi, H.; Kondo, M.; Hara-Nishimura, I.; Nishimura, M.; Machida, C.; Machida, Y. A subtilisin-like serine protease is required for epidermal surface formation in Arabidopsis embryos and juvenile plants. Development 2001, 128, 4681–4689. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.; Creff, A.; Waters, A.; Tanaka, H.; Goodrich, J.; Ingram, G.C. ZHOUPI controls embryonic cuticle formation via a signalling pathway involving the subtilisin protease ABNORMAL LEAF-SHAPE1 and the receptor kinases GASSHO1 and GASSHO2. Development 2013, 140, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.X.; Srivastava, R.; Howell, S. Overexpression of an Arabidopsis gene encoding a subtilase (AtSBT5.4) produces a clavata-like phenotype. Planta 2009, 230, 687–697. [Google Scholar] [CrossRef]
- Neuteboom, L.W.; Veth-Tello, L.M.; Clijdesdale, O.R.; Hooykaas, P.J.; van der Zaal, B.J. A novel subtilisin-like protease gene from Arabidopsis thaliana is expressed at sites of lateral root emergence. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 1999, 6, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Rautengarten, C.; Usadel, B.; Neumetzler, L.; Hartmann, J.; Büssis, D.; Altmann, T. A subtilisin-like serine protease essential for mucilage release from Arabidopsis seed coats. Plant J. Cell Mol. Biol. 2008, 54, 466–480. [Google Scholar] [CrossRef]
- Zhao, C.; Johnson, B.J.; Kositsup, B.; Beers, E.P. Exploiting secondary growth in Arabidopsis. Construction of xylem and bark cDNA libraries and cloning of three xylem endopeptidases. Plant Physiol. 2000, 123, 1185–1196. [Google Scholar] [CrossRef] [Green Version]
- Sénéchal, F.; Graff, L.; Surcouf, O.; Marcelo, P.; Rayon, C.; Bouton, S.; Mareck, A.; Mouille, G.; Stintzi, A.; Höfte, H.; et al. Arabidopsis PECTIN METHYLESTERASE17 is co-expressed with and processed by SBT3.5, a subtilisin-like serine protease. Ann. Bot. 2014, 114, 1161–1175. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.X.; Srivastava, R.; Che, P.; Howell, S.H. An endoplasmic reticulum stress response in Arabidopsis is mediated by proteolytic processing and nuclear relocation of a membrane-associated transcription factor, bZIP28. Plant Cell 2007, 19, 4111–4119. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.X.; Srivastava, R.; Che, P.; Howell, S.H. Salt stress responses in Arabidopsis utilize a signal transduction pathway related to endoplasmic reticulum stress signaling. Plant J. 2007, 51, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Ghorbani, S.; Hoogewijs, K.; Pečenková, T.; Fernandez, A.; Inzé, A.; Eeckhout, D.; Kawa, D.; De Jaeger, G.; Beeckman, T.; Madder, A.; et al. The SBT6.1 subtilase processes the GOLVEN1 peptide controlling cell elongation. J. Exp. Bot. 2016, 67, 4877–4887. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Liu, J.X.; Guo, H.; Yin, Y.; Howell, S.H. Regulation and processing of a plant peptide hormone, AtRALF23, in Arabidopsis. Plant J. 2009, 59, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, V.; López, A.; Mauch-Mani, B.; Gil, M.J.; Vera, P. An extracellular subtilase switch for immune priming in Arabidopsis. PLoS Pathog. 2013, 9, e1003445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, X.; Zhang, Z.; Wang, J.; Zuo, K. Characterization of a Novel Cotton Subtilase Gene GbSBT1 in Response to Extracellular Stimulations and Its Role in Verticillium Resistance. PLoS ONE 2016, 11, e0153988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendel, J.F.; Albert, V.A. Phylogenetics of the Cotton Genus (Gossypium): Character-State Weighted Parsimony Analysis of Chloroplast-DNA Restriction Site Data and Its Systematic and Biogeographic Implications. Syst. Bot. 1992, 17, 115–143. [Google Scholar] [CrossRef]
- Sunilkumar, G.; Campbell, L.M.; Puckhaber, L.; Stipanovic, R.D.; Rathore, K.S. Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc. Natl. Acad. Sci. USA 2006, 103, 18054–18059. [Google Scholar] [CrossRef] [Green Version]
- Wendel, J.F. Genome evolution in polyploids. Plant Mol. Biol. 2000, 42, 225–249. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Adams, K.L. Expression partitioning between genes duplicated by polyploidy under abiotic stress and during organ development. Curr. Biol. 2007, 17, 1669–1674. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Jung, S.; Cheng, C.H.; Lee, T.; Zheng, P.; Buble, K.; Crabb, J.; Humann, J.; Hough, H.; Jones, D.; et al. CottonGen: The Community Database for Cotton Genomics, Genetics, and Breeding Research. Plants 2021, 10, 2805. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Xu, W.; Zhang, K.; Zhang, L.; Yi, X.; Yao, D.; Wang, C.; Zhang, X.; Zhao, X.; Provart, N.J.; et al. ccNET: Database of co-expression networks with functional modules for diploid and polyploid Gossypium. Nucleic Acids Res. 2017, 45, D1090–D1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, L.P.; Sowdhamini, R. Cross genome comparisons of serine proteases in Arabidopsis and rice. BMC Genom. 2006, 7, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norero, N.S.; Castellote, M.A.; de la Canal, L.; Sergio, E.F. Genome-Wide Analyses of Subtilisin-Like Serine Proteases on Solanum tuberosum. Am. J. Potato Res. 2016, 93, 485–496. [Google Scholar] [CrossRef]
- Jin, X.; Liu, Y.; Hou, Z.; Zhang, Y.; Fang, Y.; Huang, Y.; Cai, H.; Qin, Y.; Cheng, Y. Genome-Wide Investigation of SBT Family Genes in Pineapple and Functional Analysis of AcoSBT1.12 in Floral Transition. Front. Genet. 2021, 12, 730821. [Google Scholar] [CrossRef]
- Cao, J.; Han, X.; Zhang, T.; Yang, Y.; Huang, J.; Hu, X. Genome-wide and molecular evolution analysis of the subtilase gene family in Vitis vinifera. BMC Genom. 2014, 15, 1116. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, J.; Costa, G.J.; Maia, M.; Paulo, O.S.; Malhó, R.; Sousa Silva, M.; Figueiredo, A. Revisiting Vitis vinifera Subtilase Gene Family: A Possible Role in Grapevine Resistance against Plasmopara viticola. Front. Plant Sci. 2016, 7, 1783. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, S.; Li, L.; Sahu, S.K.; Petersen, M.; Liu, X.; Melkonian, M.; Zhang, G.; Liu, H. Molecular evidence for origin, diversification and ancient gene duplication of plant subtilases (SBTs). Sci. Rep. 2019, 9, 12485. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kong, Y.; Chen, J.; Li, L.; Li, X.; Yu, F.; Ming, Z. Crystal structure of the extracellular domain of the receptor-like kinase TMK3 from Arabidopsis thaliana. Acta crystallographica. Sect. F Struct. Biol. Commun. 2020, 76, 384–390. [Google Scholar] [CrossRef]
- Julkowska, M.M.; Klei, K.; Fokkens, L.; Haring, M.A.; Schranz, M.E.; Testerink, C. Natural variation in rosette size under salt stress conditions corresponds to developmental differences between Arabidopsis accessions and allelic variation in the LRR-KISS gene. J. Exp. Bot. 2016, 67, 2127–2138. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, S.J.; Shin, Y.J.; Kang, J.H.; Kim, M.R.; Nam, K.H.; Lee, M.S.; Lee, S.H.; Kim, Y.H.; Hong, S.K.; et al. An atypical soybean leucine-rich repeat receptor-like kinase, GmLRK1, may be involved in the regulation of cell elongation. Planta 2009, 229, 811–821. [Google Scholar] [CrossRef]
- Clay, N.K.; Nelson, T. VH1, a provascular cell-specific receptor kinase that influences leaf cell patterns in Arabidopsis. Plant Cell 2002, 14, 2707–2722. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, R.; Gui, J.; Zhong, Y.; Li, L. The Receptor-Like Kinase AtVRLK1 Regulates Secondary Cell Wall Thickening. Plant Physiol. 2018, 177, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nsibo, D.L.; Juhar, H.M.; Govers, F.; Bouwmeester, K. Ectopic expression of Arabidopsis L-type lectin receptor kinase genes LecRK-I.9 and LecRK-IX.1 in Nicotiana benthamiana confers Phytophthora resistance. Plant Cell Rep. 2016, 35, 845–855. [Google Scholar] [CrossRef] [Green Version]
- Begum, T.; Reuter, R.; Schöffl, F. Overexpression of AtHsfB4 induces specific effects on root development of Arabidopsis. Mech. Dev. 2013, 130, 54–60. [Google Scholar] [CrossRef]
- Hernández-Coronado, M.; Ortiz-Ramírez, C. Root Patterning: Tuning SHORT ROOT Function Creates Diversity in Form. Front. Plant Sci. 2021, 12, 745861. [Google Scholar] [CrossRef]
- Moreno-Piovano, G.S.; Moreno, J.E.; Cabello, J.V.; Arce, A.L.; Otegui, M.E.; Chan, R.L. A role for LAX2 in regulating xylem development and lateral-vein symmetry in the leaf. Ann. Bot. 2017, 120, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Ranocha, P.; Dima, O.; Nagy, R.; Felten, J.; Corratgé-Faillie, C.; Novák, O.; Morreel, K.; Lacombe, B.; Martinez, Y.; Pfrunder, S.; et al. Arabidopsis WAT1 is a vacuolar auxin transport facilitator required for auxin homoeostasis. Nat. Commun. 2013, 4, 2625. [Google Scholar] [CrossRef] [Green Version]
- Prabhakaran Mariyamma, N.; Clarke, K.J.; Yu, H.; Wilton, E.E.; Van Dyk, J.; Hou, H.; Schultz, E.A. Members of the Arabidopsis FORKED1-LIKE gene family act to localize PIN1 in developing veins. J. Exp. Bot. 2018, 69, 4773–4790. [Google Scholar] [CrossRef]
- Lewis, D.R.; Olex, A.L.; Lundy, S.R.; Turkett, W.H.; Fetrow, J.S.; Muday, G.K. A kinetic analysis of the auxin transcriptome reveals cell wall remodeling proteins that modulate lateral root development in Arabidopsis. Plant Cell 2013, 25, 3329–3346. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, R.J.; Han, T.T.; Cai, W.; Fu, Z.W.; Lu, Y.T. Salt stress reduces root meristem size by nitric oxide-mediated modulation of auxin accumulation and signaling in Arabidopsis. Plant Physiol. 2015, 168, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Ribba, T.; Garrido-Vargas, F.; O’Brien, J.A. Auxin-mediated responses under salt stress: From developmental regulation to biotechnological applications. J. Exp. Bot. 2020, 71, 3843–3853. [Google Scholar] [CrossRef]
- Deslauriers, S.D.; Larsen, P.B. FERONIA is a key modulator of brassinosteroid and ethylene responsiveness in Arabidopsis hypocotyls. Mol. Plant 2010, 3, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Hématy, K.; Höfte, H. Growth control and cell wall signaling in plants. Annu. Rev. Plant Biol. 2012, 63, 381–407. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Kita, D.; Peaucelle, A.; Cartwright, H.N.; Doan, V.; Duan, Q.; Liu, M.C.; Maman, J.; Steinhorst, L.; Schmitz-Thom, I.; et al. The FERONIA Receptor Kinase Maintains Cell-Wall Integrity during Salt Stress through Ca2+ Signaling. Curr. Biol. 2018, 28, 666–675.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.S.; Lin, C.J.; Hwang, J.K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. A Publ. Protein Soc. 2004, 13, 1402–1406. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic. Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obayashi, T.; Aoki, Y.; Tadaka, S.; Kagaya, Y.; Kinoshita, K. ATTED-II in 2018: A Plant Coexpression Database Based on Investigation of the Statistical Property of the Mutual Rank Index. Plant Cell Physiol. 2018, 59, e3. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, Y.; Di, C.; Zhang, Q.; Zhang, K.; Wang, C.; You, Q.; Yan, H.; Dai, S.Y.; Yuan, J.S. JAZ7 negatively regulates dark-induced leaf senescence in Arabidopsis. J. Exp. Bot. 2016, 67, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gossypium hirsutum | Gossypium barbadense | Gossypium arboreum | Gossypium raimondii | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A Subgenome | D Subgenome | Scaffold | Total | A Subgenome | D Subgenome | Scaffold | Total | A Genome | D Genome | |
| SBT1 | 25 | 23 | 2 | 50 | 26 | 25 | 0 | 51 | 34 | 27 |
| SBT2 | 7 | 7 | 0 | 14 | 7 | 8 | 0 | 15 | 8 | 8 |
| SBT3 | 5 | 6 | 0 | 11 | 6 | 6 | 0 | 12 | 6 | 8 |
| SBT4 | 14 | 24 | 0 | 38 | 11 | 15 | 1 | 27 | 27 | 23 |
| SBT5 | 11 | 9 | 1 | 21 | 13 | 10 | 0 | 23 | 10 | 11 |
| SBT6 | 7 | 5 | 0 | 12 | 4 | 6 | 0 | 11 | 4 | 7 |
| Total | 69 | 74 | 3 | 146 | 67 | 70 | 1 | 138 | 89 | 84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, T.; Liu, L.; Zhang, X.; Li, Z.; Sheng, M.; Ge, X.; Xu, W.; Su, Z. Genome-Wide Investigation and Co-Expression Network Analysis of SBT Family Gene in Gossypium. Int. J. Mol. Sci. 2023, 24, 5760. https://doi.org/10.3390/ijms24065760
Xue T, Liu L, Zhang X, Li Z, Sheng M, Ge X, Xu W, Su Z. Genome-Wide Investigation and Co-Expression Network Analysis of SBT Family Gene in Gossypium. International Journal of Molecular Sciences. 2023; 24(6):5760. https://doi.org/10.3390/ijms24065760
Chicago/Turabian StyleXue, Tianxi, Lisen Liu, Xinyi Zhang, Zhongqiu Li, Minghao Sheng, Xiaoyang Ge, Wenying Xu, and Zhen Su. 2023. "Genome-Wide Investigation and Co-Expression Network Analysis of SBT Family Gene in Gossypium" International Journal of Molecular Sciences 24, no. 6: 5760. https://doi.org/10.3390/ijms24065760