
BCL-XL Overexpression Protects Pancreatic β-Cells against Cytokine- and Palmitate-Induced Apoptosis
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
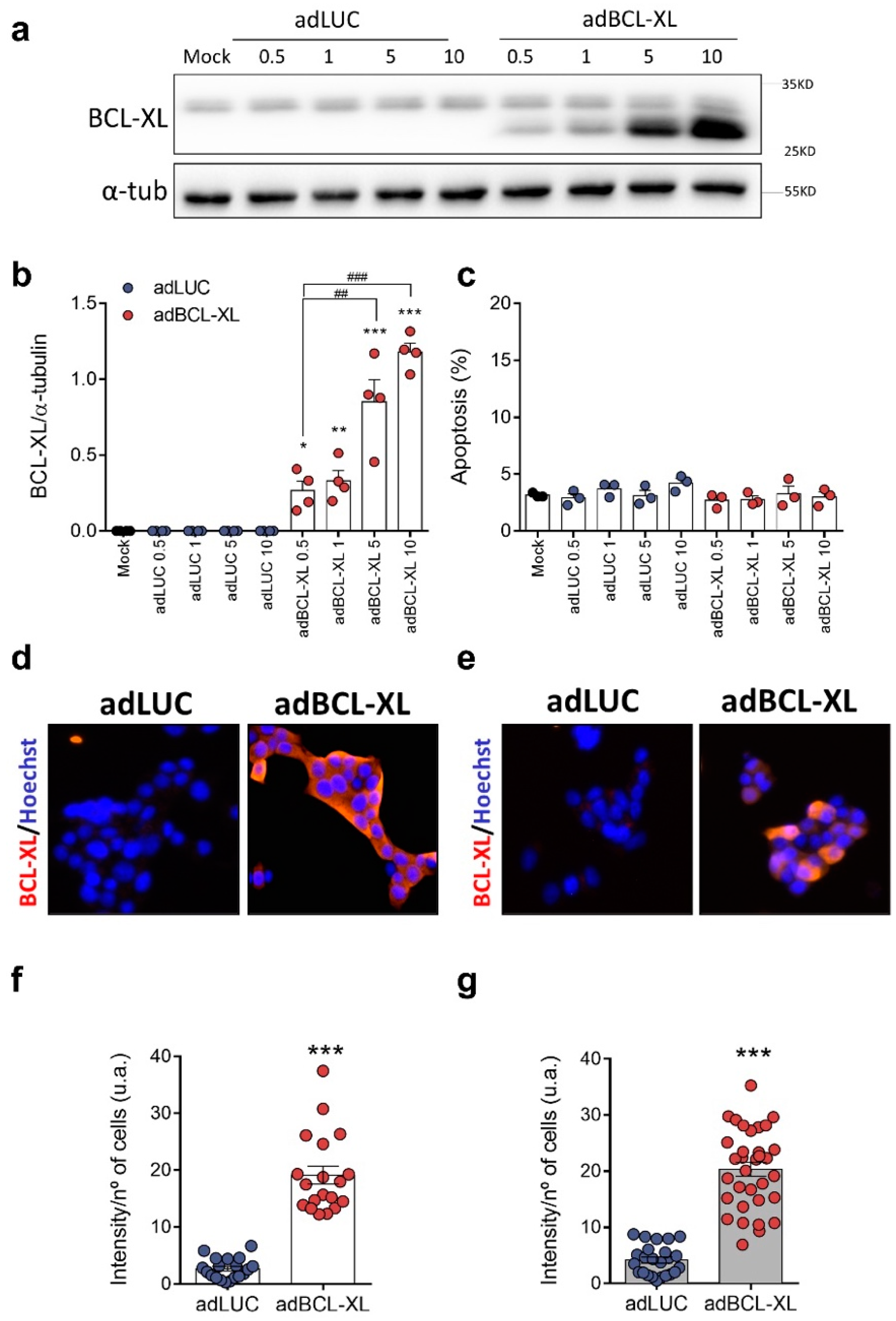
2.1. BCL-XL Overexpression in Rat and Human β-Cell Models
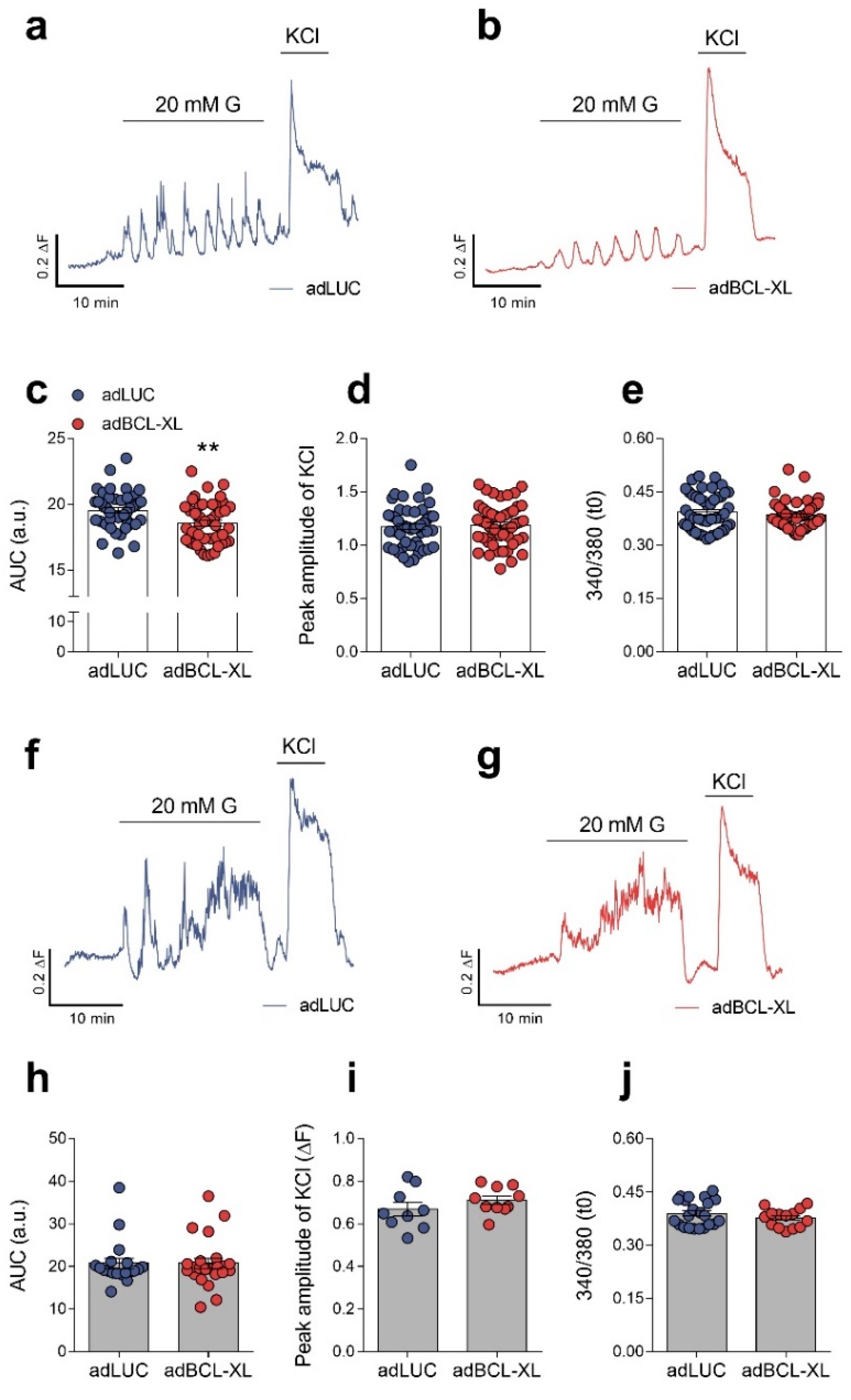
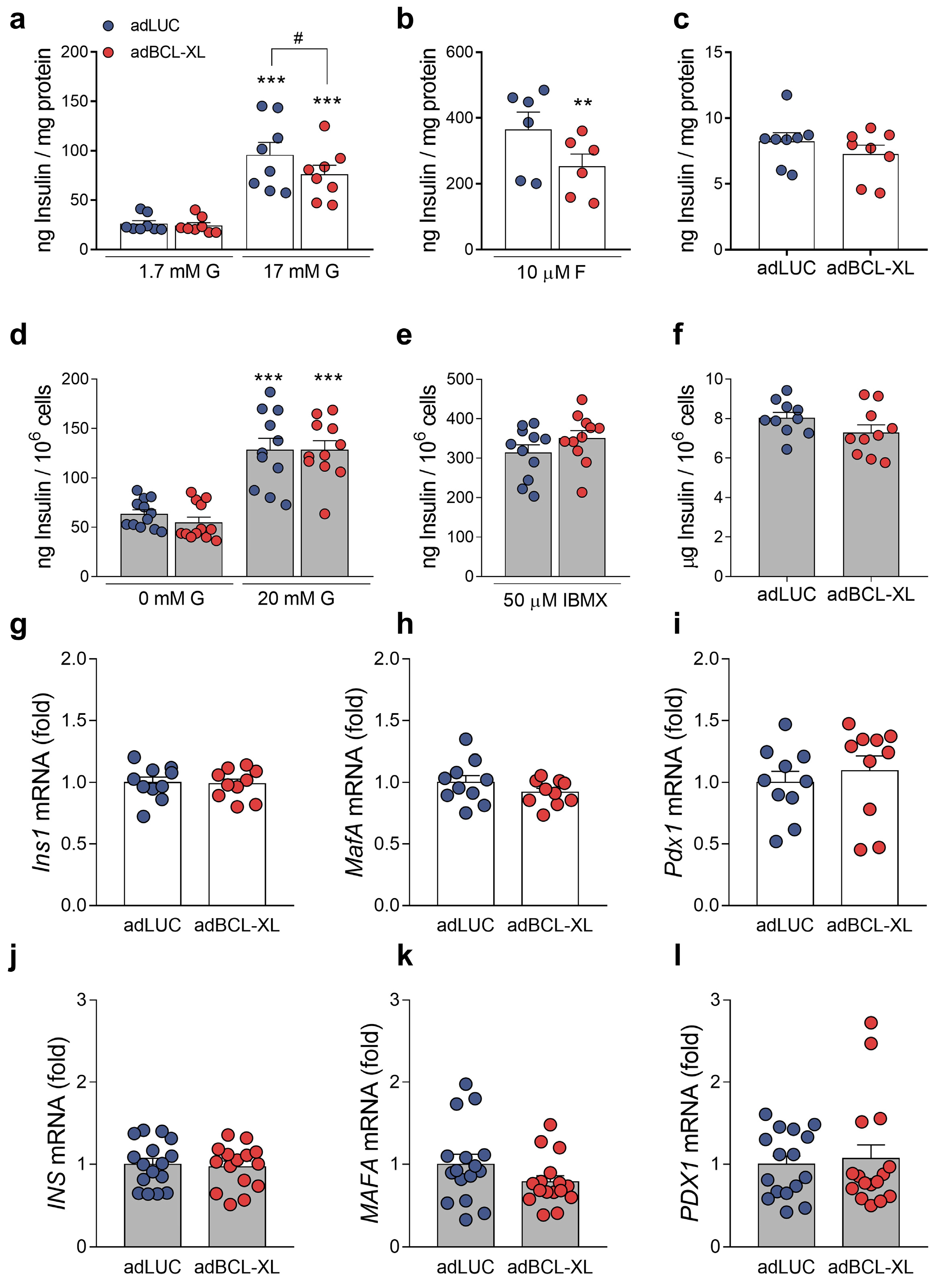
2.2. Modulation of Intracellular Ca2+ Signals and Glucose-Stimulated Insulin Secretion by BCL-XL
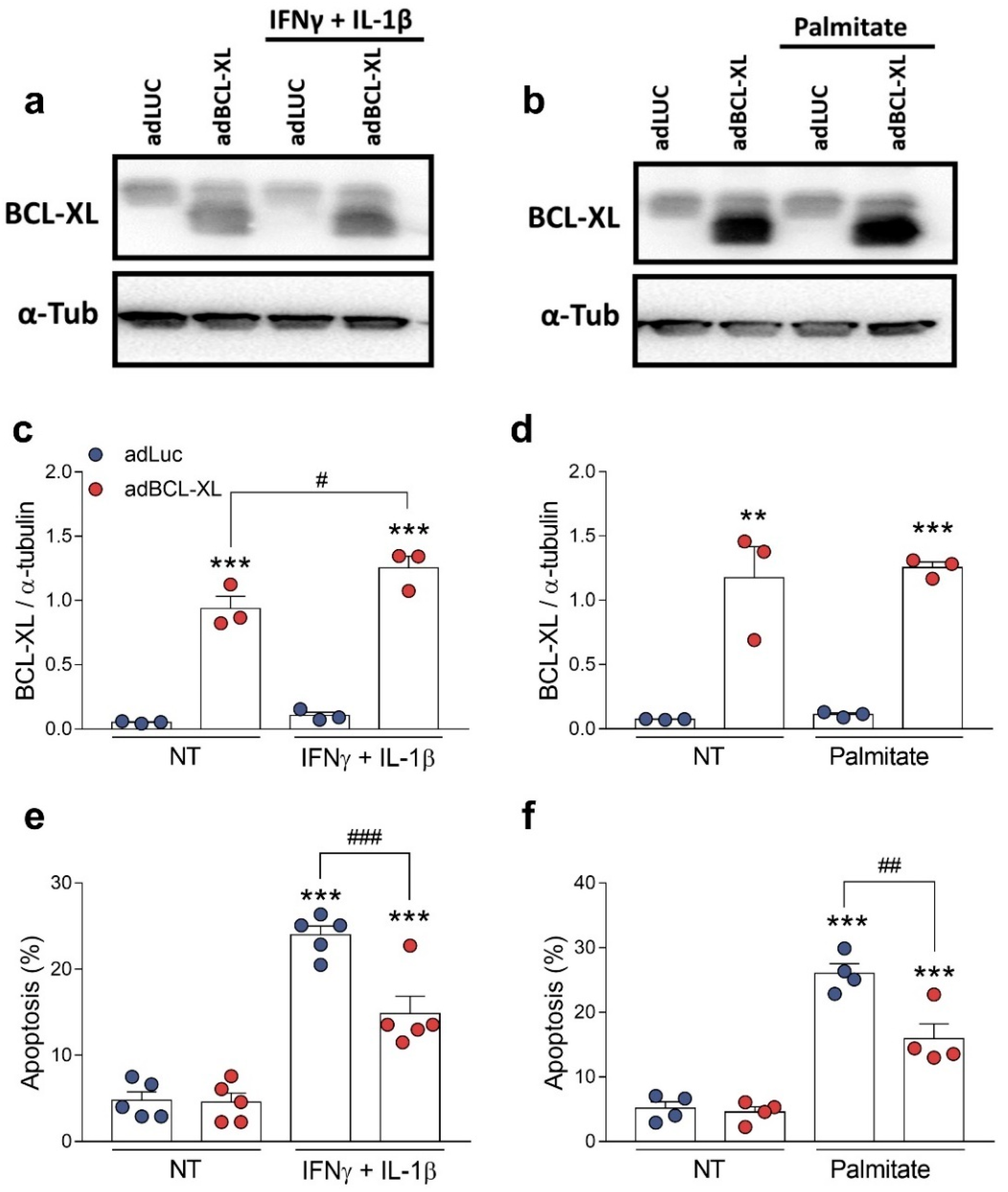
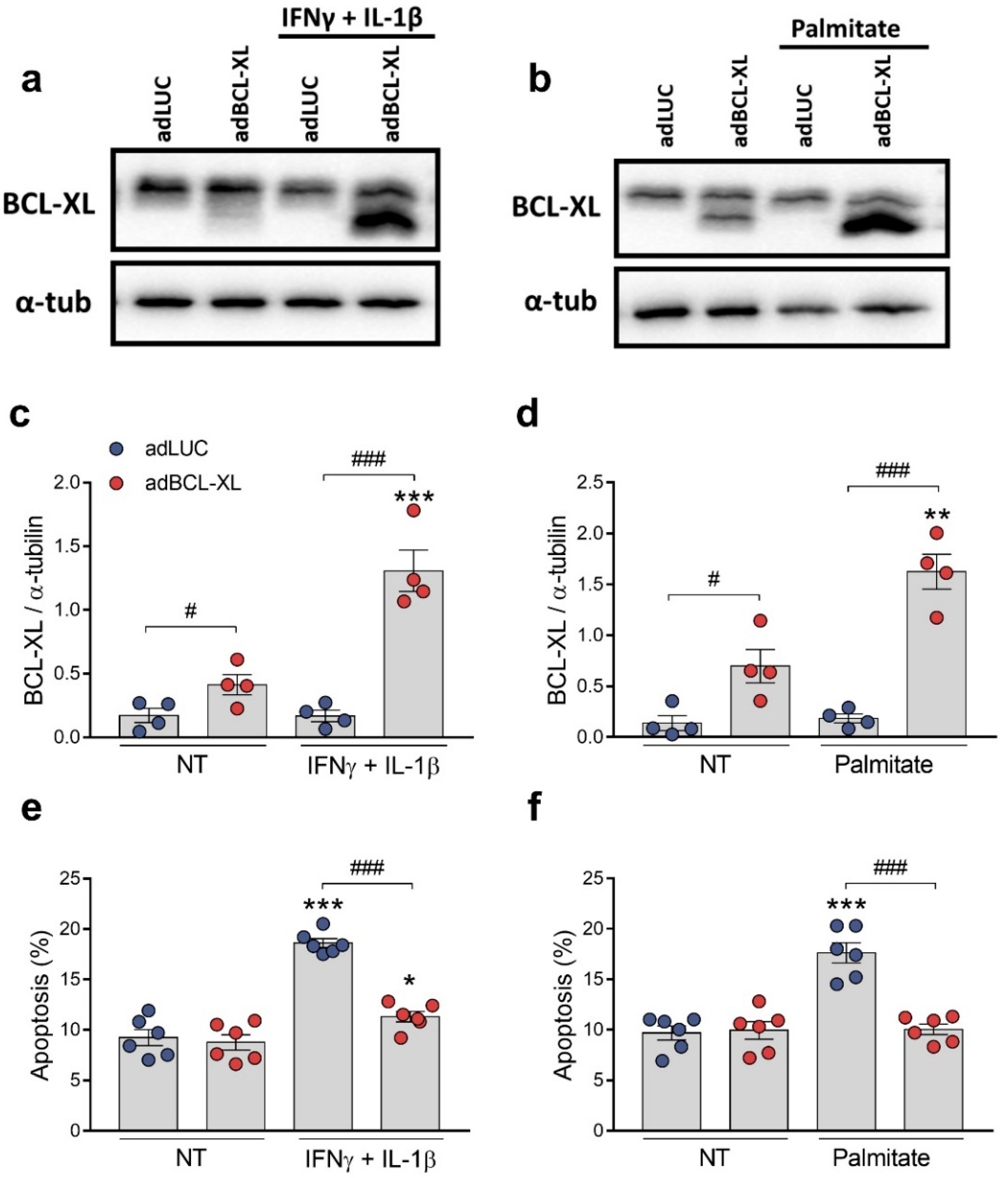
2.3. BCL-XL Overexpression Protects β-Cells against Cytokine- and Palmitate-Induced Apoptosis
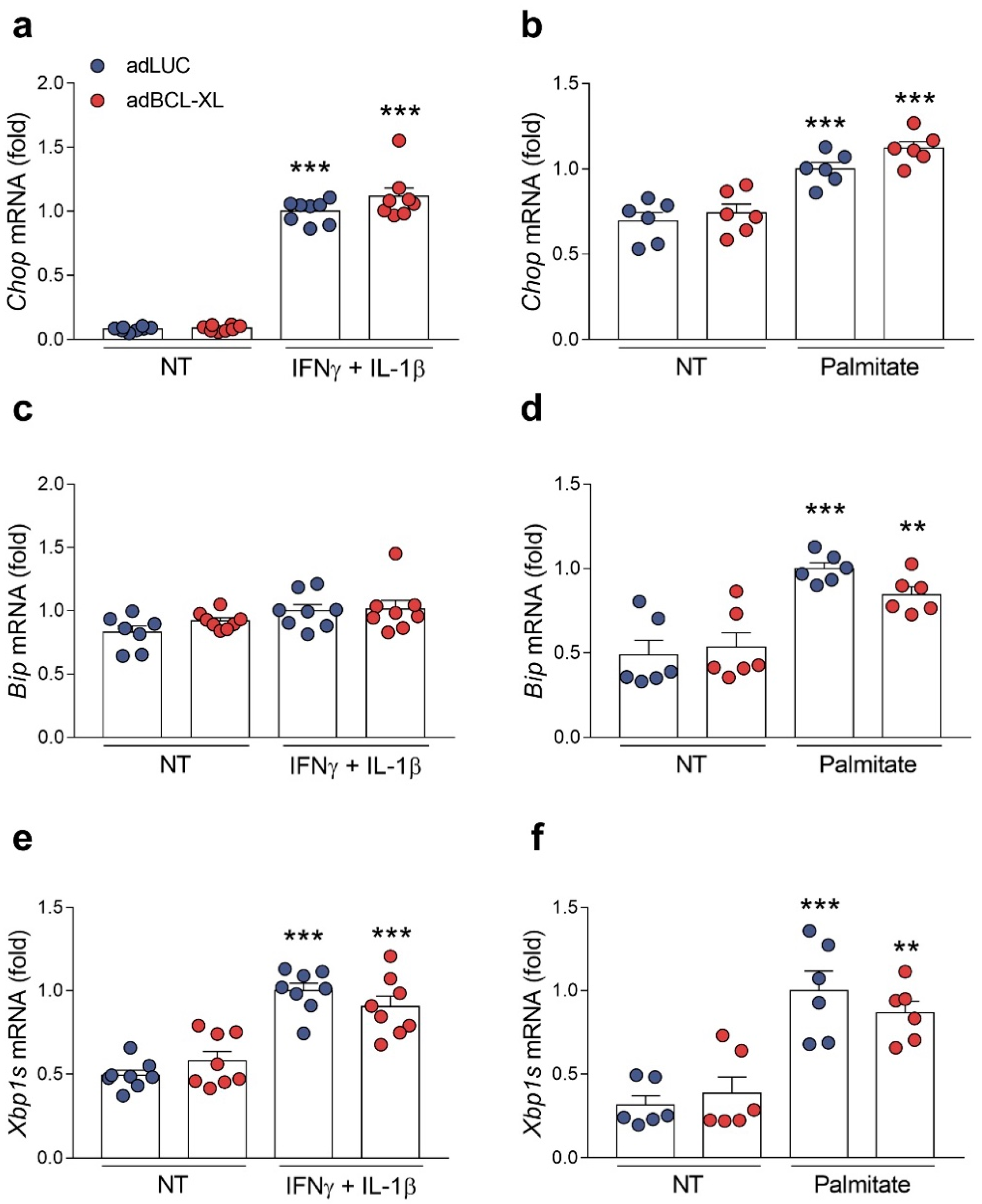
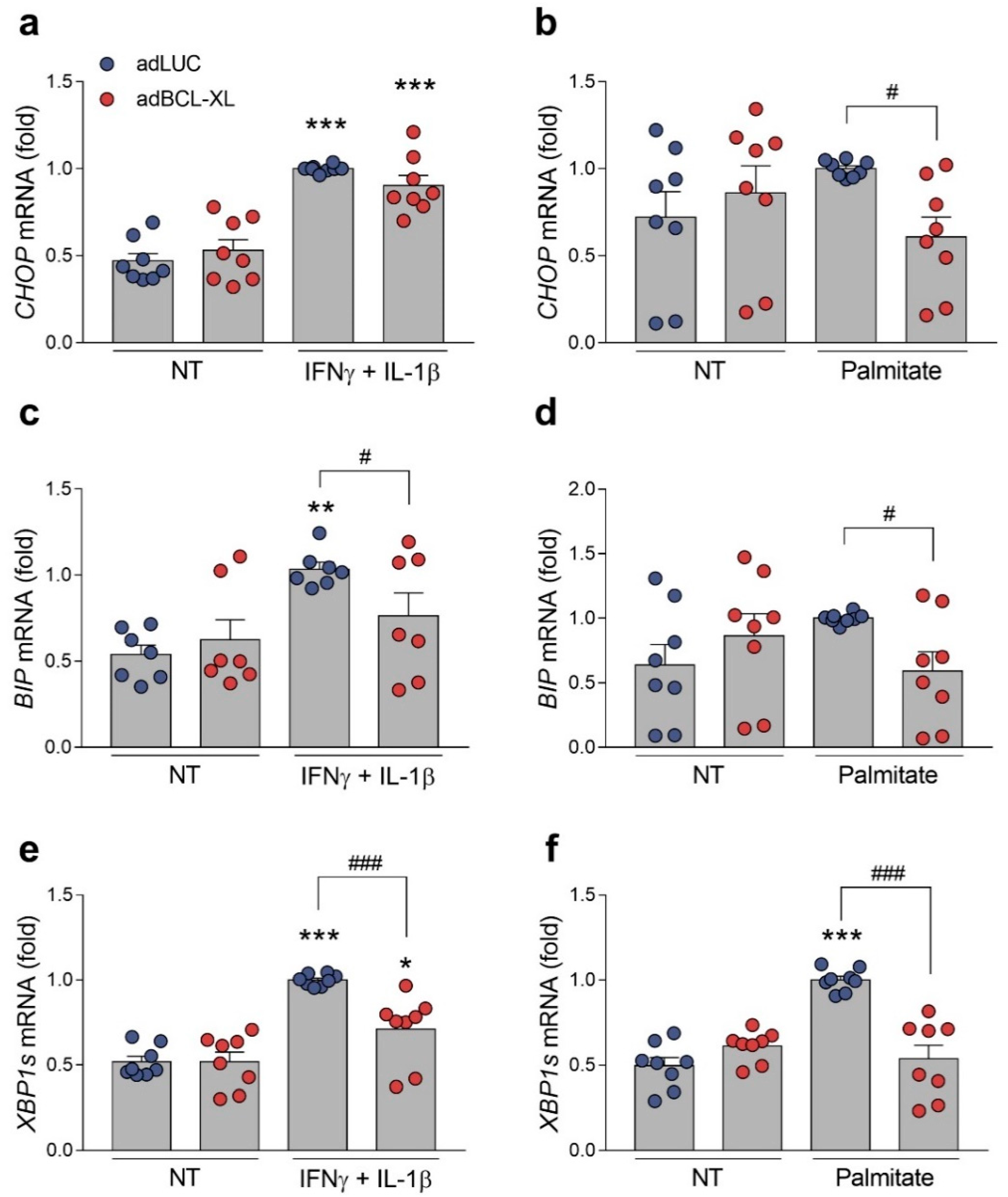
2.4. BCL-XL Overexpression Alleviates Cytokine- and Palmitate-Induced ER Stress in Human but Not in Rat β-Cells
3. Discussion
4. Materials and Methods
4.1. Culture of EndoC-βH1 and INS-1E Cells
4.2. BCL-XL Overexpression
4.3. Cell Treatments
4.4. Western Blot Analysis and Immunofluorescence
4.5. mRNA Extraction and Real-Time PCR
4.6. Intracellular Ca2+ Analysis
4.7. Glucose-Stimulated Insulin Secretion
4.8. Assessment of Cell Viability
4.9. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of Pancreatic β-Cell Death in Type 1 and Type 2 Diabetes: Many Differences, Few Similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-Cells in Type 1 and Type 2 Diabetes Mellitus: Different Pathways to Failure. Nat. Rev. Endocrinol. 2020, 26, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Eizirik, D.L. Bcl-2 Proteins in Diabetes: Mitochondrial Pathways of β-Cell Death and Dysfunction. Trends Cell Biol. 2011, 21, 424–431. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-XL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthson, J.; Germano, C.M.; Moore, F.; Maida, A.; Drucker, D.J.; Marchetti, P.; Gysemans, C.; Mathieu, C.; Nuñez, G.; Jurisicova, A.; et al. Cytokines Tumor Necrosis Factor-α and Interferon-γ Induce Pancreatic β-Cell Apoptosis through STAT1-Mediated Bim Protein Activation. J. Biol. Chem. 2011, 286, 39632–39643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurzov, E.N.; Ortis, F.; Cunha, D.A.; Gosset, G.; Li, M.; Cardozo, A.K.; Eizirik, D.L. Signaling by IL-1β+IFN-γ and ER Stress Converge on DP5/Hrk Activation: A Novel Mechanism for Pancreatic β-Cell Apoptosis. Cell Death Differ. 2009, 16, 1539–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, D.A.; Igoillo-Esteve, M.; Gurzov, E.N.; Germano, C.M.; Naamane, N.; Marhfour, I.; Fukaya, M.; Vanderwinden, J.-M.; Gysemans, C.; Mathieu, C.; et al. Death Protein 5 and P53-Upregulated Modulator of Apoptosis Mediate the Endoplasmic Reticulum Stress–Mitochondrial Dialog Triggering Lipotoxic Rodent and Human β-Cell Apoptosis. Diabetes 2012, 61, 2763–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, L.S.W.; Soetedjo, A.A.P.; Lau, H.H.; Ng, N.H.J.; Ghosh, S.; Nguyen, L.; Krishnan, V.G.; Choi, H.; Roca, X.; Hoon, S.; et al. BCL-XL/BCL2L1 Is a Critical Anti-Apoptotic Protein That Promotes the Survival of Differentiating Pancreatic Cells from Human Pluripotent Stem Cells. Cell Death Dis. 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Szegezdi, E.; Ritter, T.; O’Brien, T.; Samali, A. Cytokine-Induced β-Cell Apoptosis Is NO-Dependent, Mitochondria-Mediated and Inhibited by BCL-XL. J. Cell. Mol. Med. 2008, 12, 591–606. [Google Scholar] [CrossRef] [Green Version]
- Carrington, E.M.; McKenzie, M.D.; Jansen, E.; Myers, M.; Fynch, S.; Kos, C.; Strasser, A.; Kay, T.W.; Scott, C.L.; Allison, J. Islet β-Cells Deficient in Bcl-XL Develop but Are Abnormally Sensitive to Apoptotic Stimuli. Diabetes 2009, 58, 2316–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurzov, E.N.; Germano, C.M.; Cunha, D.A.; Ortis, F.; Vanderwinden, J.-M.; Marchetti, P.; Zhang, L.; Eizirik, D.L. P53 Up-Regulated Modulator of Apoptosis (PUMA) Activation Contributes to Pancreatic β-Cell Apoptosis Induced by Proinflammatory Cytokines and Endoplasmic Reticulum Stress. J. Biol. Chem. 2010, 285, 19910–19920. [Google Scholar] [CrossRef] [Green Version]
- Miani, M.; Barthson, J.; Colli, M.L.; Brozzi, F.; Cnop, M.; Eizirik, D.L. Endoplasmic Reticulum Stress Sensitizes Pancreatic Beta Cells to Interleukin-1β-Induced Apoptosis via Bim/A1 Imbalance. Cell Death Dis. 2013, 4, e701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.-P.; Pena, J.C.; Roe, M.W.; Mittal, A.; Levisetti, M.; Baldwin, A.C.; Pugh, W.; Ostrega, D.; Ahmed, N.; Bindokas, V.P.; et al. Overexpression of Bcl-x(L) in β-Cells Prevents Cell Death but Impairs Mitochondrial Signal for Insulin Secretion. Am. J. Physiol.-Endocrinol. Metab. 2000, 278, E340–E351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroqui, L.; Masini, M.; Merino, B.; Grieco, F.A.; Millard, I.; Dubois, C.; Quesada, I.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pancreatic α Cells Are Resistant to Metabolic Stress-Induced Apoptosis in Type 2 Diabetes. EBioMedicine 2015, 2, 378–385. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sandler, S.; Welsh, N.; Cetkovic-Cvrlje, M.; Nieman, A.; Geller, D.A.; Pipeleers, D.G.; Bendtzen, K.; Hellerström, C. Cytokines Suppress Human Islet Function Irrespective of Their Effects on Nitric Oxide Generation. J. Clin. Investig. 1994, 93, 1968–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Pipeleers, D.G.; Ling, Z.; Welsh, N.; Hellerström, C.; Andersson, A. Major Species Differences between Humans and Rodents in the Susceptibility to Pancreatic Beta-Cell Injury. Proc. Natl. Acad. Sci. USA 1994, 91, 9253–9256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines Induce Endoplasmic Reticulum Stress in Human, Rat and Mouse Beta Cells via Different Mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Deng, S.; Kucher, T.; Shaked, A.; Ketchum, R.J.; Brayman, K.L. Adenoviral Transfection of Isolated Pancreatic Islets: A Study of Programmed Cell Death (Apoptosis) and Islet Function. J. Surg. Res. 1997, 69, 23–32. [Google Scholar] [CrossRef]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The Endoplasmic Reticulum Gateway to Apoptosis by Bcl-XL Modulation of the InsP3R. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Luciani, D.S.; White, S.A.; Widenmaier, S.B.; Saran, V.V.; Taghizadeh, F.; Hu, X.; Allard, M.F.; Johnson, J.D. Bcl-2 and Bcl-XL Suppress Glucose Signaling in Pancreatic β-Cells. Diabetes 2013, 62, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilon, P.; Chae, H.-Y.; Rutter, G.A.; Ravier, M.A. Calcium Signaling in Pancreatic β-Cells in Health and in Type 2 Diabetes. Cell Calcium 2014, 56, 340–361. [Google Scholar] [CrossRef]
- Klec, C.; Ziomek, G.; Pichler, M.; Malli, R.; Graier, W.F. Calcium Signaling in SS-Cell Physiology and Pathology: A Revisit. Int. J. Mol. Sci. 2019, 20, 6110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and Execution of Lipotoxic ER Stress in Pancreatic β-Cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [Green Version]
- Janumyan, Y.M.; Sansam, C.G.; Chattopadhyay, A.; Cheng, N.; Soucie, E.L.; Penn, L.Z.; Andrews, D.; Knudson, C.M.; Yang, E. Bcl-XL/Bcl-2 Coordinately Regulates Apoptosis, Cell Cycle Arrest and Cell Cycle Entry. EMBO J. 2003, 22, 5459–5470. [Google Scholar] [CrossRef] [Green Version]
- Borrás, C.; Mas-Bargues, C.; Román-Domínguez, A.; Sanz-Ros, J.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. BCL-XL, a Mitochondrial Protein Involved in Successful Aging: From C. Elegans to Human Centenarians. Int. J. Mol. Sci. 2020, 21, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-XL Regulates Metabolic Efficiency of Neurons through Interaction with the Mitochondrial F1FO ATP Synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Soria, B.; Gauthier, B.R. Dual Trade of Bcl-2 and Bcl-XL in Islet Physiology: Balancing Life and Death With Metabolism Secretion Coupling. Diabetes 2012, 62, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.L.; Gillet, G.; Prudent, J.; Popgeorgiev, N. Bcl-2 Family of Proteins in the Control of Mitochondrial Calcium Signalling: An Old Chap with New Roles. Int. J. Mol. Sci. 2021, 22, 3730. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.-C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and Physical Interaction between Bcl-XL and a BH3-like Domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-XL Play Important Roles in the Crosstalk between Autophagy and Apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Pasula, D.J.; Shi, R.; Vanderkruk, B.; Shih, A.Z.L.; Zou, Y.; Chaudhry, A.; Hoffman, B.G.; Luciani, D.S. Bcl-XL Restricts Transcriptional, Morphological and Functional Decompensation of β-Cell Mitochondria under Chronic Glucose Excess. bioRxiv 2021. [Google Scholar] [CrossRef]
- Brun, T.; Franklin, I.; St-Onge, L.; Biason-Lauber, A.; Schoenle, E.J.; Wollheim, C.B.; Gauthier, B.R. The Diabetes-Linked Transcription Factor PAX4 Promotes β-Cell Proliferation and Survival in Rat and Human Islets. J. Cell Biol. 2004, 167, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Tsuchiya, K.; Shibata, T.; Seiwa, E. Translocation of Bim to the Endoplasmic Reticulum (ER) Mediates ER Stress Signaling for Activation of Caspase-12 during ER Stress-Induced Apoptosis. J. Biol. Chem. 2004, 279, 50375–50381. [Google Scholar] [CrossRef] [Green Version]
- Gaudette, B.T.; Iwakoshi, N.N.; Boise, L.H. Bcl-XL Protein Protects from C/EBP Homologous Protein (CHOP)-Dependent Apoptosis during Plasma Cell Differentiation. J. Biol. Chem. 2014, 289, 23629–23640. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Olberding, K.E.; White, C.; Li, C. Bcl-2 Proteins Regulate ER Membrane Permeability to Luminal Proteins during ER Stress-Induced Apoptosis. Cell Death Differ. 2011, 18, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, L.; Nguyen, T.; Gadet, R.; Lohez, O.; Mikaelian, I.; Gonzalo, P.; Luyten, T.; Chalabi-Dcha, M.; Bultynck, G.; Rimokh, R.; et al. The Endoplasmic Reticulum Pool of Bcl-XL Dampens the Unfolded Protein Response through IP3R-Dependent Calcium Release. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wyżewski, Z.; Świtlik, W.; Mielcarska, M.B.; Gregorczyk-Zboroch, K.P. The Role of Bcl-XL Protein in Viral Infections. Int. J. Mol. Sci. 2021, 22, 1956. [Google Scholar] [CrossRef]
- Ni, L.; Li, T.; Liu, B.; Song, X.; Yang, G.; Wang, L.; Miao, S.; Liu, C. The Protective Effect of Bcl-Xl Overexpression against Oxidative Stress-Induced Vascular Endothelial Cell Injury and the Role of the Akt/ENOS Pathway. Int. J. Mol. Sci. 2013, 14, 22149–22162. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Perkins, G.A.; Poblenz, A.T.; Harris, J.B.; Hung, M.; Ellisman, M.H.; Fox, D.A. Bcl-XL Overexpression Blocks Bax-Mediated Mitochondrial Contact Site Formation and Apoptosis in Rod Photoreceptors of Lead-Exposed Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1022–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, V.; Ayuso, E.; Mallol, C.; Agudo, J.; Casellas, A.; Obach, M.; Muñoz, S.; Salavert, A.; Bosch, F. In Vivo Genetic Engineering of Murine Pancreatic Beta Cells Mediated by Single-Stranded Adeno-Associated Viral Vectors of Serotypes 6, 8 and 9. Diabetologia 2011, 54, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallol, C.; Casana, E.; Jimenez, V.; Casellas, A.; Haurigot, V.; Jambrina, C.; Sacristan, V.; Morró, M.; Agudo, J.; Vilà, L.; et al. AAV-Mediated Pancreatic Overexpression of Igf1 Counteracts Progression to Autoimmune Diabetes in Mice. Mol. Metab. 2017, 6, 664–680. [Google Scholar] [CrossRef]
- Ramzy, A.; Tudurí, E.; Glavas, M.M.; Baker, R.K.; Mojibian, M.; Fox, J.K.; O’Dwyer, S.M.; Dai, D.; Hu, X.; Denroche, H.C.; et al. AAV8 Ins1-Cre Can Produce Efficient β-Cell Recombination but Requires Consideration of off-Target Effects. Sci. Rep. 2020, 10, 10518. [Google Scholar] [CrossRef]
- Singh, K.; Bricard, O.; Haughton, J.; Björkqvist, M.; Thorstensson, M.; Luo, Z.; Mascali, L.; Pasciuto, E.; Mathieu, C.; Dooley, J.; et al. Gene Delivery of Manf to Beta-Cells of the Pancreatic Islets Protects NOD Mice from Type 1 Diabetes Development. Biomolecules 2022, 12, 1493. [Google Scholar] [CrossRef]
- Pena, J.C.; Rudin, C.M.; Thompson, C.B. A Bcl-XL Transgene Promotes Malignant Conversion of Chemically Initiated Skin Papillomas. Cancer Res. 1998, 58, 2111–2116. [Google Scholar]
- Ramírez-Komo, J.A.; Delaney, M.A.; Straign, D.; Lukin, K.; Tsang, M.; Iritani, B.M.; Hagman, J. Spontaneous Loss of B Lineage Transcription Factors Leads to Pre-B Leukemia in Ebf1+/–Bcl-XLTg Mice. Oncogenesis 2017, 6, e355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donahue, R.J.; Fehrman, R.L.; Gustafson, J.R.; Nickells, R.W. BCLXL Gene Therapy Moderates Neuropathology in the DBA/2J Mouse Model of Inherited Glaucoma. Cell Death Dis. 2021, 12, 781. [Google Scholar] [CrossRef] [PubMed]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A Genetically Engineered Human Pancreatic β Cell Line Exhibiting Glucose-Inducible Insulin Secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef]
- Santin, I.; Dos Santos, R.S.; Eizirik, D.L. Pancreatic Beta Cell Survival and Signaling Pathways: Effects of Type 1 Diabetes-Associated Genetic Variants. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1433, pp. 21–54. [Google Scholar]
- Dos Santos, R.S.; Marroqui, L.; Grieco, F.A.; Marselli, L.; Suleiman, M.; Henz, S.R.; Marchetti, P.; Wernersson, R.; Eizirik, D.L. Protective Role of Complement C3 against Cytokine-Mediated β-Cell Apoptosis. Endocrinology 2017, 158, 2503–2521. [Google Scholar] [CrossRef]
- Marroqui, L.; Santin, I.; Dos Santos, R.S.; Marselli, L.; Marchetti, P.; Eizirik, D.L. BACH2, a Candidate Risk Gene for Type 1 Diabetes, Regulates Apoptosis in Pancreatic β-Cells via JNK1 Modulation and Crosstalk with the Candidate Gene PTPN2. Diabetes 2014, 63, 2516–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Mandrup-Poulsen, T. A Choice of Death—The Signal-Transduction of Immune-Mediated Beta-Cell Apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Kutlu, B.; Cardozo, A.K.; Darville, M.I.; Kruhøffer, M.; Magnusson, N.; Ørntoft, T.; Eizirik, D.L. Discovery of Gene Networks Regulating Cytokine-Induced Dysfunction and Apoptosis in Insulin-Producing INS-1 Cells. Diabetes 2003, 52, 2701–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortis, F.; Cardozo, A.K.; Crispim, D.; Störling, J.; Mandrup-Poulsen, T.; Eizirik, D.L. Cytokine-Induced Proapoptotic Gene Expression in Insulin-Producing Cells Is Related to Rapid, Sustained, and Nonoscillatory Nuclear Factor-ΚB Activation. Mol. Endocrinol. 2006, 20, 1867–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.F.; Cunha, D.A.; Ladriere, L.; Igoillo-Esteve, M.; Bugliani, M.; Marchetti, P.; Cnop, M. In Vitro Use of Free Fatty Acids Bound to Albumin: A Comparison of Protocols. BioTechniques 2015, 58, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovskii, C.; Kristinsson, H.; Elksnis, A.; Wang, X.; Gavali, H.; Bergsten, P.; Scharfmann, R.; Welsh, N. EndoC-ΒH1 Cells Display Increased Sensitivity to Sodium Palmitate When Cultured in DMEM/F12 Medium. Islets 2017, 9, e1296995. [Google Scholar] [CrossRef] [Green Version]
- Babiloni-Chust, I.; dos Santos, R.S.; Medina-Gali, R.M.; Perez-Serna, A.A.; Encinar, J.-A.; Martinez-Pinna, J.; Gustafsson, J.-A.; Marroqui, L.; Nadal, A. G Protein-Coupled Estrogen Receptor Activation by Bisphenol-A Disrupts the Protection from Apoptosis Conferred by the Estrogen Receptors ERα and ERβ in Pancreatic Beta Cells. Environ. Int. 2022, 164, 107250. [Google Scholar] [CrossRef]
- Marroqui, L.; Dos Santos, R.S.; Fløyel, T.; Grieco, F.A.; Santin, I.; Op De Beeck, A.; Marselli, L.; Marchetti, P.; Pociot, F.; Eizirik, D.L. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic β-Cells. Diabetes 2015, 64, 3808–3817. [Google Scholar] [CrossRef] [Green Version]
- Marroqui, L.; Dos Santos, R.S.; Op de beeck, A.; Coomans de Brachène, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α Mediates Human Beta Cell HLA Class I Overexpression, Endoplasmic Reticulum Stress and Apoptosis, Three Hallmarks of Early Human Type 1 Diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Villar-Pazos, S.; Martinez-Pinna, J.; Castellano-Muñoz, M.; Alonso-Magdalena, P.; Marroqui, L.; Quesada, I.; Gustafsson, J.A.; Nadal, A. Molecular Mechanisms Involved in the Non-Monotonic Effect of Bisphenol-A on Ca2+ Entry in Mouse Pancreatic β-Cells. Sci. Rep. 2017, 7, 11770. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gonzalez, A.; Merino, B.; Marroquí, L.; Ñeco, P.; Alonso-Magdalena, P.; Caballero-Garrido, E.; Vieira, E.; Soriano, S.; Gomis, R.; Nadal, A.; et al. Insulin Hypersecretion in Islets from Diet-Induced Hyperinsulinemic Obese Female Mice Is Associated with Several Functional Adaptations in Individual β-Cells. Endocrinology 2013, 154, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.S.; Medina-Gali, R.M.; Babiloni-Chust, I.; Marroqui, L.; Nadal, A. In Vitro Assays to Identify Metabolism-Disrupting Chemicals with Diabetogenic Activity in a Human Pancreatic β-Cell Model. Int. J. Mol. Sci. 2022, 23, 5040. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.S.; Daures, M.; Philippi, A.; Romero, S.; Marselli, L.; Marchetti, P.; Senée, V.; Bacq, D.; Besse, C.; Baz, B.; et al. DUTPase (DUT) Is Mutated in a Novel Monogenic Syndrome with Diabetes and Bone Marrow Failure. Diabetes 2017, 66, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Hoorens, A.; Van De Casteele, M.; Klöppel, G.; Pipeleers, D. Glucose Promotes Survival of Rat Pancreatic β Cells by Activating Synthesis of Proteins Which Suppress a Constitutive Apoptotic Program. J. Clin. Investig. 1996, 98, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.; Colli, M.L.; Cnop, M.; Esteve, M.I.; Cardozo, A.K.; Cunha, D.A.; Bugliani, M.; Marchetti, P.; Eizirik, D.L. PTPN2, a Candidate Gene for Type 1 Diabetes, Modulates Interferon-γ- Induced Pancreatic β-Cell Apoptosis. Diabetes 2009, 58, 1283–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allagnat, F.; Fukaya, M.; Nogueira, T.C.; Delaroche, D.; Welsh, N.; Marselli, L.; Marchetti, P.; Haefliger, J.A.; Eizirik, D.L.; Cardozo, A.K. C/EBP Homologous Protein Contributes to Cytokine-Induced pro-Inflammatory Responses and Apoptosis in β-Cells. Cell Death Differ. 2012, 19, 1836–1846. [Google Scholar] [CrossRef]
- Dos Santos, R.S.; Marroqui, L.; Velayos, T.; Olazagoitia-Garmendia, A.; Jauregi-Miguel, A.; Castellanos-Rubio, A.; Eizirik, D.L.; Castaño, L.; Santin, I. DEXI, a Candidate Gene for Type 1 Diabetes, Modulates Rat and Human Pancreatic Beta Cell Inflammation via Regulation of the Type I IFN/STAT Signalling Pathway. Diabetologia 2019, 62, 459–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, F.; Santin, I.; Nogueira, T.C.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. The Transcription Factor C/EBP Delta Has Anti-Apoptotic and Anti-Inflammatory Roles in Pancreatic Beta Cells. PLoS ONE 2012, 7, e31062. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Serna, A.A.; Dos Santos, R.S.; Ripoll, C.; Nadal, A.; Eizirik, D.L.; Marroqui, L. BCL-XL Overexpression Protects Pancreatic β-Cells against Cytokine- and Palmitate-Induced Apoptosis. Int. J. Mol. Sci. 2023, 24, 5657. https://doi.org/10.3390/ijms24065657
Perez-Serna AA, Dos Santos RS, Ripoll C, Nadal A, Eizirik DL, Marroqui L. BCL-XL Overexpression Protects Pancreatic β-Cells against Cytokine- and Palmitate-Induced Apoptosis. International Journal of Molecular Sciences. 2023; 24(6):5657. https://doi.org/10.3390/ijms24065657
Chicago/Turabian StylePerez-Serna, Atenea A., Reinaldo S. Dos Santos, Cristina Ripoll, Angel Nadal, Decio L. Eizirik, and Laura Marroqui. 2023. "BCL-XL Overexpression Protects Pancreatic β-Cells against Cytokine- and Palmitate-Induced Apoptosis" International Journal of Molecular Sciences 24, no. 6: 5657. https://doi.org/10.3390/ijms24065657