

A Case of Mucopolysaccharidosis II Caused by a Novel Variant with Skin Linear Hyperpigmented Streaks along Blaschko’s Lines
 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
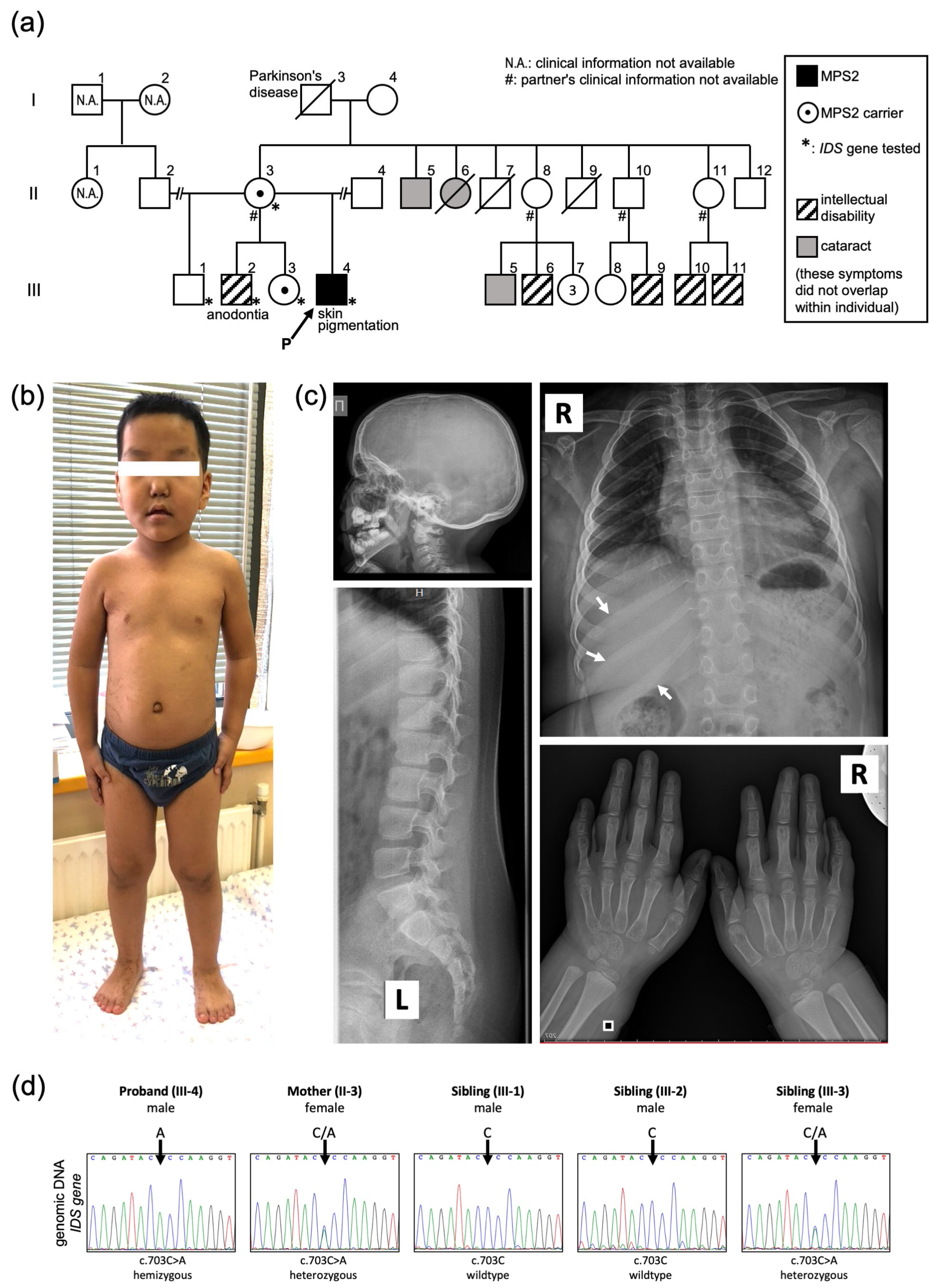
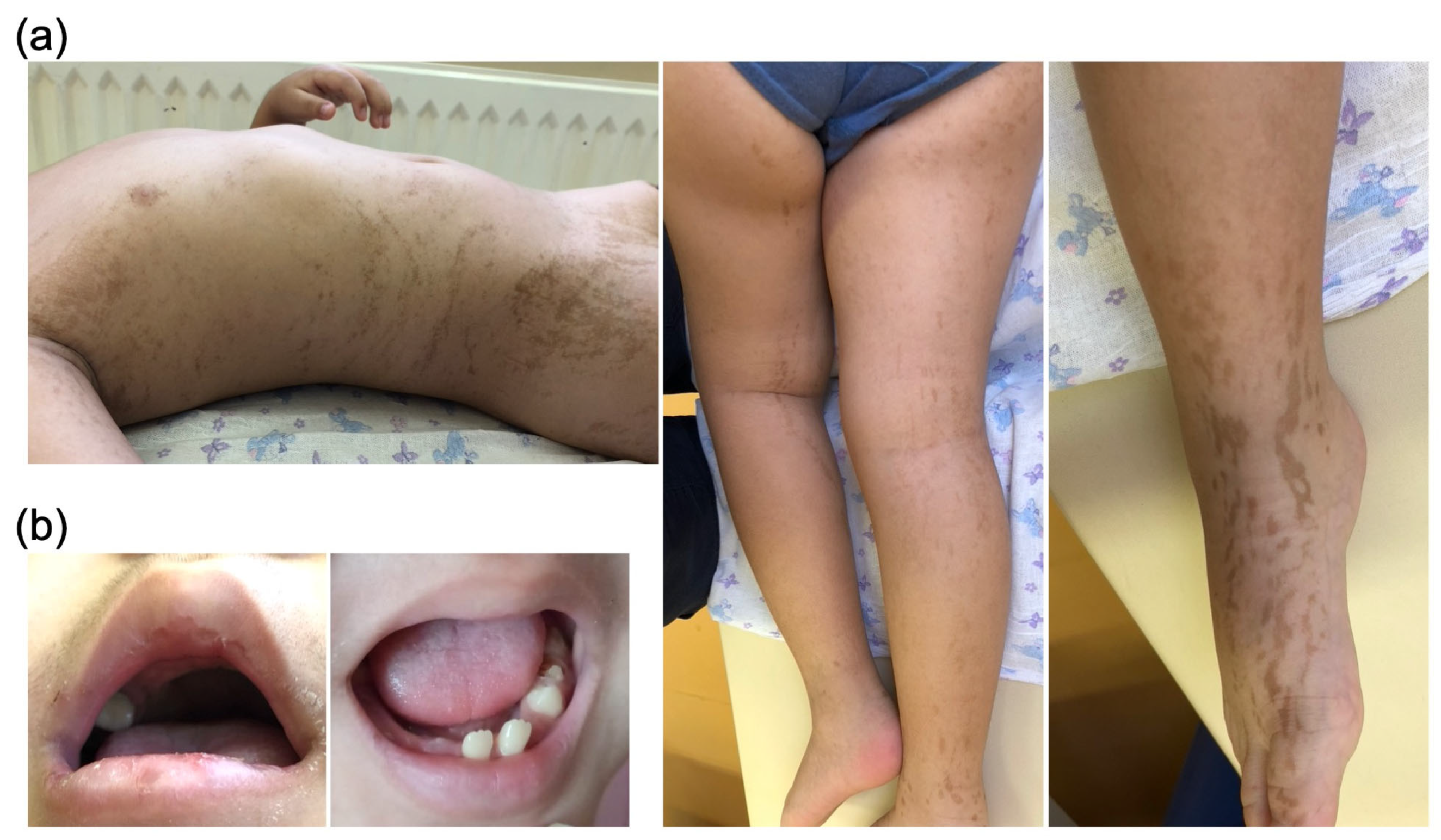
Case Presentation
3. Discussion
4. Materials and Methods
Molecular Diagnosis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neufeld, N.F.; Muenzer, J. The Mucopolysaccharidoses. The Online Metabolic and Molecular Bases of Inherited Disease. 2019. Available online: https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225544161 (accessed on 22 October 2022).
- Bondeson, M.L.; Dahl, N.; Malmgren, H.; Kleijer, W.J.; Tönnesen, T.; Carlberg, B.M.; Pettersson, U. Inversion of the IDS gene resulting from recombination with IDS-related sequences is a common cause of the Hunter syndrome. Hum. Mol. Genet. 1995, 4, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M. Mucopolysaccharidosis Type II. GeneReviews® [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1274/ (accessed on 22 October 2022).
- Burton, B.K.; Giugliani, R. Diagnosing Hunter syndrome in pediatric practice: Practical considerations and common pitfalls. Eur. J. Pediatr. 2012, 171, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Gene Mutations Database at the Institute of Medical Genetics in Cardiff. 2003. Available online: https://www.hgmd.cf.ac.uk/ac/index.php (accessed on 22 October 2022).
- Ochiai, T.; Suzuki, Y.; Kato, T.; Shichino, H.; Chin, M.; Mugishima, H.; Orii, T. Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): A survey among 52 Japanese patients. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Thappa, D.M.; Singh, A.; Jaisankar, T.J.; Rao, R.; Ratnakar, C. Pebbling of the skin: A marker of Hunter’s syndrome. Pediatr. Dermatol. 1998, 15, 370–373. [Google Scholar] [CrossRef]
- Demitsu, T.; Kakurai, M.; Okubo, Y.; Shibayama, C.; Kikuchi, Y.; Mori, Y.; Sukegawa, K.; Mizuguchi, M. Skin eruption as the presenting sign of Hunter syndrome IIB. Clin. Exp. Dermatol. 1999, 24, 179–182. [Google Scholar] [CrossRef]
- Filimonenko, J.; Timofeev, V. Rukovodstvo k metodike issledovanija intellekta u detej D. Vekslera (WISC): Adapt. variant (Guide to D. Wechsler Method of Intelligence Investigation of Children), 3rd ed.; Gosstandart Rossii GP “IMATON”: St. Petersburg, Russia, 1994. (In Russian) [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Landon, G.; Denjoy, I.; Clero, E.; Silenok, A.; Kurnosova, I.; Butsenin, A.; Gourmelon, P.; Jourdain, J.R. Reference values of electrographic and cardiac ultrasound parameters in Russian healthy children and adolescents. Sci. Rep. 2021, 11, 2916. [Google Scholar] [CrossRef]
- Zoghbi, W.A.; Adams, D.; Bonow, R.O.; Enriquez-Sarano, M.; Foster, E.; Grayburn, P.A.; Hahn, R.T.; Han, Y.; Hung, J.; Lang, R.M.; et al. Recommendations for Noninvasive Evaluation of Native Valvular Regurgitation: A Report from the American Society of Echocardiography Developed in Collaboration with the Society for Cardiovascular Magnetic Resonance. J. Am. Soc. Echocardiogr. 2017, 30, 303–371. [Google Scholar] [CrossRef]
- Happle, R. Lyonization and the lines of Blaschko. Hum. Genet. 1985, 70, 200–206. [Google Scholar] [CrossRef]
- Sun, B.K.; Tsao, H. X-chromosome inactivation and skin disease. J. Investig. Dermatol. 2008, 128, 2753–2759. [Google Scholar] [CrossRef] [Green Version]
- Scheuerle, A.E.; Ursini, M.V. Incontinentia Pigmenti. GeneReviews® [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1472/ (accessed on 22 October 2022).
- Fusco, F.; Pescatore, A.; Steffann, J.; Bonnefont, J.P.; De Oliveira, J.; Lioi, M.B.; Ursini, M.V. Clinical utility gene card: For incontinentia pigmenti. Eur. J. Hum. Genet. 2019, 27, 1894–1900. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.N.; Ohtsubo, M.; Nishina, S.; Nakao, S.; Yoshida, K.; Hosono, K.; Kurata, K.; Ohishi, K.; Fukami, M.; Sato, M.; et al. Analysis of IKBKG/NEMO gene in five Japanese cases of incontinentia pigmenti with retinopathy: Fine genomic assay of a rare male case with mosaicism. J. Hum. Genet. 2021, 66, 205–214. [Google Scholar] [CrossRef]
- Fusco, F.; Fimiani, G.; Tadini, G.; Michele, D.; Ursini, M.V. Clinical diagnosis of incontinentia pigmenti in a cohort of male patients. J. Am. Acad. Dermatol. 2007, 56, 264–267. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Thomson, P.; Mutch, S.; Webster, A.R.; Rai, H.; Hill, V.; Moore, A.T. Somatic mosaicism of a novel IKBKG mutation in a male patient with incontinentia pigmenti. Am. J. Med. Genet. A. 2015, 167, 1601–1604. [Google Scholar] [CrossRef]
- Kenwrick, S.; Woffendin, H.; Jakins, T.; Shuttleworth, S.G.; Mayer, E.; Greenhalgh, L.; Whittaker, J.; Rugolotto, S.; Bardaro, T.; Esposito, T.; et al. II Consortium, Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am. J. Hum. Genet. 2001, 69, 1210–1217. [Google Scholar] [CrossRef] [Green Version]
- Pescatore, A.; Spinosa, E.; Casale, C.; Lioi, M.B.; Ursini, M.V.; Fusco, F. Human Genetic Diseases Linked to the Absence of NEMO: An Obligatory Somatic Mosaic Disorder in Male. Int. J. Mol. Sci. 2022, 23, 1179. [Google Scholar] [CrossRef]
- Diaz, J.; Berger, S.; Leon, E. TFE3-associated neurodevelopmental disorder: A distinct recognizable syndrome. Am. J. Med. Genet. A 2020, 182, 584–590. [Google Scholar] [CrossRef]
- Lehalle, D.; Vabres, P.; Sorlin, A.; Bierhals, T.; Avila, M.; Carmignac, V.; Chevarin, M.; Torti, E.; Abe, Y.; Bartolomaeus, T.; et al. De novo mutations in the X-linked TFE3 gene cause intellectual disability with pigmentary mosaicism and storage disorder-like features. J. Med. Genet. 2020, 57, 808–819. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Li, H.; Puertollano, R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. Life. Sci. 2014, 71, 2483–2497. [Google Scholar] [CrossRef] [Green Version]
- Kalter, D.C.; Griffiths, W.A.; Atherton, D.J. Linear and whorled nevoid hypermelanosis. J. Am. Acad. Dermatol. 1988, 6, 1037–1044. [Google Scholar] [CrossRef]
- Alrobaee, A.A.; Alsaif, F. Linear and whorled nevoid hypermelanosis associated with developmental delay and generalized convulsions. Int. J. Dermatol. 2004, 43, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Kromann, A.B.; Ousager, L.B.; Ali, I.K.M.; Aydemir, N.; Bygum, A. Pigmentary mosaicism: A review of original literature and recommendations for future handling. Orphanet J. Rare Dis. 2018, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, A.; Hofmann, U.B.; Hoehn, H.; Broecker, E.B.; Hamm, H. Postnatal confirmation of prenatally diagnosed trisomy 20 mosaicism in a patient with linear and whorled nevoid hypermelanosis. Pediatr. Dermatol. 2004, 21, 636–641. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease (OMIM) | MPS II (#309900) [1,3] | IP (#308300) [15] | MRXSPF (#301066) [22,23] |
|---|---|---|---|
| Gene | IDS | IKBKG/NEMO | TFE3 |
| Locus | Xq28 | Xq28 | Xp11.23 |
| Inheritance | X-linked recessive | X-linked dominant | X-linked |
| Dermatological | Ivory-colored skin lesions Extensive blue nevus (Mongolian spots) | Skin lesions (stages I-IV): I. Erythema II. Verrucous lesions III.Hyperpigmented streaks and whorls (Blaschko’s line) IV. Pale, hairless, atrophic linear streaks | Blaschkoid pigmentary |
| Nervous | Intellectual disability Developmental delay Behavior difficulties | Intellectual disability CNS vasculopathy Seizures | Intellectual disability Developmental delay Epilepsy |
| Skeletal | Dysostosis multiplex (J-shaped sella turcica, bullet-shaped phalanges, metacarpal pointing, metaphyseal widening of the long bones with cortical thinning, oar-shaped ribs, flared iliac wings, round- or hooked-shape vertebral bodies) Joint contracture Hip dysplasia | N.D. | Thickening of ribs Hyperlordosis Scoliosis Hip dislocation Limitation of elbow extension |
| Facial dysmorphism | Prominent forehead Flat nasal bridge Flared nostrils Synophrys Hypertelorism Thick lips Macroglossia | N.D. | Flat nasal bridge Short nose with anteverted nares Widely spaced and almond-shaped eyes Thick lips Facial hypertrichosis |
| Odontologic | Irregular shaped teeth Overgrown gingival tissue | Anodontia Microdontia Abnormally shaped teeth Delayed eruption or impaction | N.D. |
| Ophthalmological | Optic nerve atrophy Retinopathy | Strabismus Microphthalmia Retinal detachments Cataracts Optic atrophy Retinal pigmentation | Strabismus Retinal degeneration Depigmented macula of the iris Oculomotor apraxia |
| Cardiovascular | Valvular disease Cardiomyopathy Rhythm disorder | N.D. | Congenital heart defect |
| Gastrointestinal | Hepatosplenomegaly Umbilical/inguinal hernia Chronic diarrhea | N.D. | Umbilical hernia Anteriorly displaced anus |
| Laboratory test | Hypersecretion of glycosaminoglycan in urine * Low/absent enzyme activity of iduronate 2-sulfatase | N.D. | N.D. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sofronova, V.; Gurinova, E.; Petukhova, D.; Fukamatsu, H.; Yamamoto, T.; Aoyama, Y.; Golikova, P.; Moskvitin, G.; Ivanova, R.; Savvina, M.; et al. A Case of Mucopolysaccharidosis II Caused by a Novel Variant with Skin Linear Hyperpigmented Streaks along Blaschko’s Lines. Int. J. Mol. Sci. 2023, 24, 5647. https://doi.org/10.3390/ijms24065647
Sofronova V, Gurinova E, Petukhova D, Fukamatsu H, Yamamoto T, Aoyama Y, Golikova P, Moskvitin G, Ivanova R, Savvina M, et al. A Case of Mucopolysaccharidosis II Caused by a Novel Variant with Skin Linear Hyperpigmented Streaks along Blaschko’s Lines. International Journal of Molecular Sciences. 2023; 24(6):5647. https://doi.org/10.3390/ijms24065647
Chicago/Turabian StyleSofronova, Viktoriia, Elizaveta Gurinova, Diana Petukhova, Hiroko Fukamatsu, Takenobu Yamamoto, Yumi Aoyama, Polina Golikova, Gavril Moskvitin, Roza Ivanova, Mira Savvina, and et al. 2023. "A Case of Mucopolysaccharidosis II Caused by a Novel Variant with Skin Linear Hyperpigmented Streaks along Blaschko’s Lines" International Journal of Molecular Sciences 24, no. 6: 5647. https://doi.org/10.3390/ijms24065647